Effets de xénobiotiques sur la reproduction
2011
| ANALYSE |
14-
Toxicocinétique
Du point de vue du toxicologue, le parcours d’un xénobiotique dans l’organisme peut se schématiser selon quatre étapes majeures (figure 14.1 ). La première correspond à l’absorption, c’est-à-dire le passage du milieu extérieur vers le milieu intérieur. La seconde est la distribution du composé dans les différents compartiments de l’organisme, les compartiments se traduisant par un ou plusieurs sites de stockage, des organes ou des tissus cibles, et des sites de biotransformation de la substance absorbée (3e étape). Enfin, l’étape d’élimination consiste à rejeter dans le milieu extérieur le xénobiotique préalablement absorbé.
). La première correspond à l’absorption, c’est-à-dire le passage du milieu extérieur vers le milieu intérieur. La seconde est la distribution du composé dans les différents compartiments de l’organisme, les compartiments se traduisant par un ou plusieurs sites de stockage, des organes ou des tissus cibles, et des sites de biotransformation de la substance absorbée (3e étape). Enfin, l’étape d’élimination consiste à rejeter dans le milieu extérieur le xénobiotique préalablement absorbé.
). La première correspond à l’absorption, c’est-à-dire le passage du milieu extérieur vers le milieu intérieur. La seconde est la distribution du composé dans les différents compartiments de l’organisme, les compartiments se traduisant par un ou plusieurs sites de stockage, des organes ou des tissus cibles, et des sites de biotransformation de la substance absorbée (3e étape). Enfin, l’étape d’élimination consiste à rejeter dans le milieu extérieur le xénobiotique préalablement absorbé.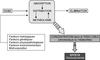

Certes, ce schéma est simplifié et de nombreux xénobiotiques peuvent être stockés dans différents tissus ou subir des modifications chimiques dans plusieurs organes, mais il reflète la réalité de deux processus que l’on a coutume de distinguer mais qui sont intimement liés : la toxicocinétique et la toxicodynamie. La toxicocinétique couvre les phénomènes d’absorption, de distribution tissulaire, de métabolisme et d’excrétion, c’est-à-dire qu’elle a pour principal objectif de déterminer la quantité de substance toxique susceptible d’atteindre sa cible et de préciser sous quelle forme (composé initial ou métabolites) elle y parvient. La toxicodynamie se préoccupe de l’interaction du xénobiotique avec sa cible et de l’effet toxique que cela produit. Les interactions entre ces deux domaines déterminent la toxicité d’une substance. De façon générale, plus la forme toxique d’un xénobiotique est capable d’atteindre la cible et plus cette dernière est sensible à l’agent exogène et plus l’effet toxique sera important.
Dans la mesure où la toxicocinétique est un élément critique de la caractérisation du danger d’une substance chimique, il convient de la prendre en compte dans toute évaluation du risque. Dans ce chapitre, nous traiterons des différentes phases prises en compte dans les approches de toxicocinétique telles qu’elles sont définies dans la ligne directrice 417 de l’OCDE qui vient d’être adoptée (OECD, 2010). Comme il est souligné dans ce document, les données toxicocinétiques peuvent aider à évaluer l’adéquation et la pertinence des données de toxicité animale, à des fins d’extrapolation des dangers pour l’homme, et peuvent apporter des informations utiles pour déterminer les effets liés à la voie d’administration, la biodisponibilité et autres aspects relatifs à la conception de l’étude. En outre, les données sur le métabolisme peuvent fournir des informations utiles pour évaluer l’importance de la prise en compte sur le plan toxicologique, de l’exposition à des métabolites exogènes du xénobiotique. Des données toxicocinétiques adéquates aideront à confirmer l’acceptabilité et l’applicabilité des méthodes fondées sur les relations quantitatives structure-activité et les prévisions établies à partir de données croisées sur des substances analogues. Les données toxicocinétiques peuvent également servir à évaluer la pertinence toxicologique d’autres études et par exemple aider à l’extrapolation in vivo/in vitro.
). Comme il est souligné dans ce document, les données toxicocinétiques peuvent aider à évaluer l’adéquation et la pertinence des données de toxicité animale, à des fins d’extrapolation des dangers pour l’homme, et peuvent apporter des informations utiles pour déterminer les effets liés à la voie d’administration, la biodisponibilité et autres aspects relatifs à la conception de l’étude. En outre, les données sur le métabolisme peuvent fournir des informations utiles pour évaluer l’importance de la prise en compte sur le plan toxicologique, de l’exposition à des métabolites exogènes du xénobiotique. Des données toxicocinétiques adéquates aideront à confirmer l’acceptabilité et l’applicabilité des méthodes fondées sur les relations quantitatives structure-activité et les prévisions établies à partir de données croisées sur des substances analogues. Les données toxicocinétiques peuvent également servir à évaluer la pertinence toxicologique d’autres études et par exemple aider à l’extrapolation in vivo/in vitro.Les exemples chargés d’illustrer les différentes parties seront principalement issus de données relatives aux substances perturbatrices endocriniennes reprotoxiques.
Absorption
L’absorption est le processus par lequel une substance pénètre dans l’organisme. Deux éléments de ce processus sont importants à prendre en compte : la quantité absorbée et la vitesse d’absorption. Bien qu’il existe des différences liées à la barrière à franchir (peau, poumon, paroi intestinale), l’absorption des petites molécules revient pour la plupart d’entre elles à un mécanisme de franchissement passif d’une ou plusieurs membranes (Lehman-McKeeman, 2008). Ce phénomène dépend donc en premier lieu des propriétés physico-chimiques de la substance elle-même, c’est-à-dire sa masse moléculaire, son degré d’ionisation, sa réactivité, sa solubilité. Les agents chimiques lipophiles sont mieux à même de traverser une membrane dont les constituants sont pour l’essentiel des lipides. En revanche, les molécules ionisées ont très peu de chances de franchir cet obstacle sous cette forme, sauf pour les très petites molécules qui peuvent dans certains cas diffuser au niveau des pores membranaires. Il existe généralement une bonne corrélation entre le caractère lipophile d’un xénobiotique et son degré d’absorption. Toutefois, en particulier au niveau de l’intestin, cette relation n’augmente plus, ou même décroît pour les substances très lipophiles (log Kow1
> 4), en raison de la difficulté à former une solution ou une émulsion dans la lumière intestinale (Walsh et coll., 1997). Ainsi, l’absorption digestive chez le rat du BDE 153, un hexabromodiphényléther qui a un log Kow = 6,4 (ATSDR 2004), est d’environ 75 % de la dose (Sanders et coll., 2006 ; Hakk et coll., 2009), alors que pour une dose équivalente et un log Kow = 8,6 (ATSDR 2004), le décabromodiphényléther (BDE 209) n’est absorbé qu’à environ 25 % (Riu et coll., 2008). La relation entre la dose externe et interne dépend donc en grande partie du niveau d’absorption, qui peut être lui-même affecté par le caractère lipophile de la substance ou encore, pour certains composés, de l’efficacité des systèmes de pompe à efflux tels que les P-glycoprotéines.
). Ce phénomène dépend donc en premier lieu des propriétés physico-chimiques de la substance elle-même, c’est-à-dire sa masse moléculaire, son degré d’ionisation, sa réactivité, sa solubilité. Les agents chimiques lipophiles sont mieux à même de traverser une membrane dont les constituants sont pour l’essentiel des lipides. En revanche, les molécules ionisées ont très peu de chances de franchir cet obstacle sous cette forme, sauf pour les très petites molécules qui peuvent dans certains cas diffuser au niveau des pores membranaires. Il existe généralement une bonne corrélation entre le caractère lipophile d’un xénobiotique et son degré d’absorption. Toutefois, en particulier au niveau de l’intestin, cette relation n’augmente plus, ou même décroît pour les substances très lipophiles (log Kow1
> 4), en raison de la difficulté à former une solution ou une émulsion dans la lumière intestinale (Walsh et coll., 1997). Ainsi, l’absorption digestive chez le rat du BDE 153, un hexabromodiphényléther qui a un log Kow = 6,4 (ATSDR 2004), est d’environ 75 % de la dose (Sanders et coll., 2006 ; Hakk et coll., 2009), alors que pour une dose équivalente et un log Kow = 8,6 (ATSDR 2004), le décabromodiphényléther (BDE 209) n’est absorbé qu’à environ 25 % (Riu et coll., 2008). La relation entre la dose externe et interne dépend donc en grande partie du niveau d’absorption, qui peut être lui-même affecté par le caractère lipophile de la substance ou encore, pour certains composés, de l’efficacité des systèmes de pompe à efflux tels que les P-glycoprotéines.Il faut signaler que biodisponibilité et absorption ne sont pas synonymes. La différence, par exemple, entre l’absorption orale (c’est-à-dire la présence dans la paroi intestinale et la veine porte) et la biodisponibilité (c’est-à-dire la présence dans le sang systémique et dans les tissus) peut, entre autres facteurs, provenir de la dégradation chimique liée au métabolisme de la paroi intestinale, à l’efflux vers la lumière intestinale ou encore au métabolisme présystémique dans le foie (OECD, 2010).
).Distribution
La distribution tissulaire est le processus selon lequel une substance absorbée (ou ses métabolites) se répartissent dans les différents organes et tissus. Bien que certaines barrières membranaires soient moins perméables que d’autres, les mécanismes de diffusion obéissent, de façon générale aux mêmes règles que celles qui régissent l’absorption et dépendent en premier lieu des caractéristiques physico-chimiques du xénobiotique. Cependant, en matière de perturbation endocrinienne, le franchissement de certaines barrières telles que la barrière placentaire est à examiner avec attention car il peut conduire à une exposition du fœtus pendant une période particulièrement sensible du développement. Si l’exposition du fœtus aux œstrogènes maternels est limitée en raison de leur liaison à l’α fœtoprotéine, nombre de perturbateurs endocriniens sont beaucoup moins affins à cette protéine et se retrouvent de ce fait facilement dans la circulation fœtale. C’est ce que rapportent Ikezuki et coll. (2002) et Vandenberg et coll. (2007)à propos du bisphénol A.
) et Vandenberg et coll. (2007)à propos du bisphénol A.Métabolisme et élimination
Si les mécanismes d’absorption et de transfert dans les tissus favorisent le passage des molécules non ionisées lipophiles, l’élimination de ces dernières implique au contraire qu’elles soient hydrosolubles afin de pouvoir être excrétées dans l’urine ou dans la bile. Dans la plupart des cas, cela nécessite la transformation chimique du xénobiotique qui a pénétré dans l’organisme et ces réactions sont essentiellement catalysées par des enzymes qui fonctionnent avec des co-facteurs endogènes. Conceptuellement, ce processus a été séparé en deux phases au cours desquelles le xénobiotique est oxydé, réduit ou hydrolysé (phase I) et/ou conjugué à l’acide glucuronique, à un groupement sulfate ou acétate, au glutathion ou encore à un acide aminé (phase II). Ces réactions prennent en charge non seulement les xénobiotiques, mais également les composés endogènes comme les stéroïdes, les prostaglandines ou encore certaines vitamines (figure 14.2).
).

Les monooxygénases à cytochrome P450 (CYP) jouent un rôle majeur dans le métabolisme des xénobiotiques. Ce sont des enzymes membranaires localisées dans le réticulum endoplasmique. On dénombre une soixantaine de CYP différents chez l’homme et une douzaine d’entre eux sont utilisés dans le métabolisme des xénobiotiques, parmi lesquels les formes 1A1/2, 2C9, 2C19, 2D6 et 3A4 sont les plus fréquemment impliquées (Guengerich, 2008). Ces différentes formes peuvent être inhibées, activées ou induites par les xénobiotiques, ce qui affecte non seulement le temps de séjour de la substance dans l’organisme, mais peut également se traduire par une modification des taux de synthèse ou de dégradation de certaines hormones lorsqu’elles sont prises en charge par ces enzymes. C’est ainsi que l’on explique, par exemple, l’effet féminisant de l’atrazine, herbicide longtemps utilisé dans la culture du maïs. Ce pesticide est un inducteur du CYP2C19 (aromatase), favorisant une surproduction d’œstrogènes et des effets féminisants (Holloway et coll., 2008). Il existe un polymorphisme génétique important pour plusieurs CYP, en particulier les formes 2C9, 2C19 et 2D6, conduisant à des niveaux d’expression enzymatiques très différents selon les individus (Genguerich, 2008). Ce polymorphisme se traduit par des différences interindividuelles de susceptibilité à l’action des toxiques (Hatagima, 2002).
). Ces différentes formes peuvent être inhibées, activées ou induites par les xénobiotiques, ce qui affecte non seulement le temps de séjour de la substance dans l’organisme, mais peut également se traduire par une modification des taux de synthèse ou de dégradation de certaines hormones lorsqu’elles sont prises en charge par ces enzymes. C’est ainsi que l’on explique, par exemple, l’effet féminisant de l’atrazine, herbicide longtemps utilisé dans la culture du maïs. Ce pesticide est un inducteur du CYP2C19 (aromatase), favorisant une surproduction d’œstrogènes et des effets féminisants (Holloway et coll., 2008). Il existe un polymorphisme génétique important pour plusieurs CYP, en particulier les formes 2C9, 2C19 et 2D6, conduisant à des niveaux d’expression enzymatiques très différents selon les individus (Genguerich, 2008). Ce polymorphisme se traduit par des différences interindividuelles de susceptibilité à l’action des toxiques (Hatagima, 2002).Les réactions de phase II les plus fréquemment utilisées par l’organisme sont la glucuronidation et la conjugaison au glutathion et au groupement sulfate. Alors que la conjugaison à l’acide glucuronique et au sulfate intervient sur des substrats nucléophiles, celle faisant intervenir le glutathion concerne les molécules électrophiles. L’uridine-diphosphate glucuronosyl transférase (UGT) est une famille d’enzymes capable de catalyser le transfert d’un acide glucuronique sur une molécule possédant une fonction alcool, amine ou thiol. Il existe près d’une vingtaine d’UGT différentes chez l’homme (Shipkova et Wieland, 2005), localisées dans la membrane du réticulum endoplasmique. La plupart des substrats pris en charge par les UGT peuvent également être métabolisés par les sulfotransférases qui, pour celles impliquées dans le métabolisme des xénobiotiques, sont des enzymes cytosoliques qui utilisent le phospho-adénosine-phosphosulfate comme co-facteur. Chez les vertébrés, les UGT, comme les sulfotransférases sont impliquées dans le métabolisme des hormones stéroïdiennes. Les glutathion-transférases sont des enzymes homo- ou hétéro-dimériques qui sont principalement cytosoliques mais qui peuvent, pour certaines classes, être membranaires. Les conjugués au glutathion peuvent être éliminés sous cette forme dans la bile, ou subir d’autres étapes de biotransformation conduisant à la formation d’un conjugué à la cystéine, lui-même pris en charge par les acétyltransférases pour former un conjugué mercapturique finalement éliminé dans l’urine.
), localisées dans la membrane du réticulum endoplasmique. La plupart des substrats pris en charge par les UGT peuvent également être métabolisés par les sulfotransférases qui, pour celles impliquées dans le métabolisme des xénobiotiques, sont des enzymes cytosoliques qui utilisent le phospho-adénosine-phosphosulfate comme co-facteur. Chez les vertébrés, les UGT, comme les sulfotransférases sont impliquées dans le métabolisme des hormones stéroïdiennes. Les glutathion-transférases sont des enzymes homo- ou hétéro-dimériques qui sont principalement cytosoliques mais qui peuvent, pour certaines classes, être membranaires. Les conjugués au glutathion peuvent être éliminés sous cette forme dans la bile, ou subir d’autres étapes de biotransformation conduisant à la formation d’un conjugué à la cystéine, lui-même pris en charge par les acétyltransférases pour former un conjugué mercapturique finalement éliminé dans l’urine.Les enzymes de phase II peuvent être induites ou inhibées par des xénobiotiques, mais dans une moindre mesure que les cytochromes P450. L’effet des xénobiotiques sur ces enzymes est un des mécanismes impliqués dans la perturbation endocrinienne. Il a ainsi été montré que les polychlorobiphényles (PCB), les polybromodiphényléthers (PBDE) ou les dérivés halogénés du bisphénol A, qui sont trois familles de perturbateurs endocriniens ayant des propriétés œstrogéniques, ont une affinité limitée pour ER (récepteurs aux œstrogènes), mais qu’en revanche, ils (ou leurs métabolites) ont une puissante capacité à inhiber la sulfotransférase 1E1, enzyme chargée d’inactiver l’œstradiol (Kester et coll., 2002). La même observation a été faite pour les alkylphénols et les phtalates (Waring et coll., 2008). Les récepteurs AhR, CAR et PXR jouent un rôle prépondérant dans l’induction des enzymes de phase I et II. AhR, qui est activé par les dioxines et les hydrocarbures aromatiques polyhalogénés ou non, est surtout impliqué dans l’induction des CYP1A et 1B ainsi que des UGT1A1, 1A3, 1A4 et 1A6 et de la sulfotransférase 1A1 (Jana et Paliwal, 2007 ; Tolson et Wang, 2010). L’activation des récepteurs CAR et PXR provoque l’induction des CYP 2A, 2B, 2C, 3A, de l’UGT1A1, de la sulfotransférase 2A1 et de la GST α1. CYP1A est également régulé par CAR, alors que PXR induit spécifiquement CYP7A (Tolson et Wang, 2010).
). La même observation a été faite pour les alkylphénols et les phtalates (Waring et coll., 2008). Les récepteurs AhR, CAR et PXR jouent un rôle prépondérant dans l’induction des enzymes de phase I et II. AhR, qui est activé par les dioxines et les hydrocarbures aromatiques polyhalogénés ou non, est surtout impliqué dans l’induction des CYP1A et 1B ainsi que des UGT1A1, 1A3, 1A4 et 1A6 et de la sulfotransférase 1A1 (Jana et Paliwal, 2007 ; Tolson et Wang, 2010). L’activation des récepteurs CAR et PXR provoque l’induction des CYP 2A, 2B, 2C, 3A, de l’UGT1A1, de la sulfotransférase 2A1 et de la GST α1. CYP1A est également régulé par CAR, alors que PXR induit spécifiquement CYP7A (Tolson et Wang, 2010).Même si un grand nombre de tissus peuvent exprimer des enzymes de biotransformation, c’est le foie qui est l’organe principal du métabolisme. Il est toutefois possible que certaines isoformes particulières soient spécifiquement exprimées dans un tissu extra-hépatique. C’est par exemple le cas de l’aromatase qui chez l’adulte est exprimée dans les ovaires, le placenta, le tissu adipeux, l’os ou dans une moindre mesure, le testicule, mais pas dans le foie, alors que les niveaux d’expression sont très élevés dans le foie fœtal (Simpson et coll., 2002).
).Métabolites et effets toxiques
Si ces réactions de phase I et II jouent un rôle essentiel dans l’inactivation des composés exogènes, elles sont également à l’origine des effets toxiques de plusieurs substances (Dekant, 2009). En effet, le processus de biotransformation peut donner lieu à des métabolites réactifs capables de se lier de façon irréversible à des macromolécules endogènes telles que l’ADN ou les protéines et entraîner des effets génotoxiques ou immunotoxiques (figure 14.3).
). En effet, le processus de biotransformation peut donner lieu à des métabolites réactifs capables de se lier de façon irréversible à des macromolécules endogènes telles que l’ADN ou les protéines et entraîner des effets génotoxiques ou immunotoxiques (figure 14.3).

La toxicité d’hydrocarbures aromatiques polycycliques comme le benzo(a) pyrène ou le diméthylbenzanthracène qui sont métabolisés dans un premier temps par les P450 relève de ce phénomène et conduit aux effets cancérigènes de ces polluants. Pour ce qui concerne les mécanismes de perturbation endocrinienne, les voies de bioactivation ne passent pas nécessairement par la formation d’un intermédiaire réactif capable de se lier de façon covalente à une molécule endogène, mais le plus souvent par la production d’un métabolite ayant une affinité ou une activité bien plus forte que la substance initiale vis-à-vis d’une protéine de transport ou d’un récepteur nucléaire. Kitamura et coll. (2008) ont ainsi identifié une trentaine de substances qualifiées de pro-œstrogènes parmi les composés naturels ou les contaminants. Ces substances, inactives dans leur état initial, nécessitent l’intervention d’une enzyme, généralement un P450, pour devenir œstrogéniques. Parmi les exemples cités figurent des pesticides (biphényle, DDT, métoxychlore, bifénox), des produits industriels (benzophénone, phtalates, PCB, PBDE, oligomères du styrène, diphényle méthane et diphényle propane), des polluants (B(a)P, 2-nitrofluorène), des phyto-œstrogènes (chalcone).
) ont ainsi identifié une trentaine de substances qualifiées de pro-œstrogènes parmi les composés naturels ou les contaminants. Ces substances, inactives dans leur état initial, nécessitent l’intervention d’une enzyme, généralement un P450, pour devenir œstrogéniques. Parmi les exemples cités figurent des pesticides (biphényle, DDT, métoxychlore, bifénox), des produits industriels (benzophénone, phtalates, PCB, PBDE, oligomères du styrène, diphényle méthane et diphényle propane), des polluants (B(a)P, 2-nitrofluorène), des phyto-œstrogènes (chalcone).Une fois formés, les métabolites sont excrétés dans l’urine par le rein ou éliminés dans les fèces via la bile. L’excrétion dans le lait maternel peut également intervenir de façon substantielle, comme cela a été montré pour les PBDE, les phtalates ou le bisphénol A. Généralement, les rongeurs excrètent davantage de métabolites par voie biliaire que le chien, le singe ou l’homme. Cela est dû à des différences entre espèces dans le seuil d’excrétion biliaire des métabolites. Ce seuil, qui dépend de la masse moléculaire du métabolite est d’environ 250 Da chez le rat, alors qu’il est de 400-600 Da pour le chien et l’homme (Dybing et coll., 2002). Après avoir été éliminés dans la bile, les conjugués glucuronides et sulfates peuvent facilement être hydrolysés dans le tube digestif. Les produits de biotransformation ainsi libérés sont ensuite réabsorbés par l’intestin et de nouveau métabolisés au niveau du foie. On parle alors de cycle entéro-hépatique, dont la conséquence première est une augmentation du temps de séjour du xénobiotique dans l’organisme.
). Après avoir été éliminés dans la bile, les conjugués glucuronides et sulfates peuvent facilement être hydrolysés dans le tube digestif. Les produits de biotransformation ainsi libérés sont ensuite réabsorbés par l’intestin et de nouveau métabolisés au niveau du foie. On parle alors de cycle entéro-hépatique, dont la conséquence première est une augmentation du temps de séjour du xénobiotique dans l’organisme.Des modèles plus complexes
Le terme « toxicocinétique » prend en compte la répartition du xénobiotique dans les différents compartiments de l’organisme au cours du temps. Le passage de la circulation générale vers les organes se fait généralement par diffusion passive et de ce fait la concentration du composé dans la circulation générale reflète la concentration dans le tissu cible. La relation entre dose externe et dose interne ne peut être obtenue à partir d’études traditionnelles qui mesurent l’absorption, la distribution, le métabolisme et l’excrétion du composé et de ses métabolites dans l’urine et les fèces. Elle repose sur la connaissance de l’évolution au cours du temps des concentrations plasmatiques et urinaires des substances que l’on souhaite étudier. À partir des concentrations mesurées, ces analyses permettent, par des modèles compartimentaux, de déterminer plusieurs variables telles que la clairance, la biodisponibilité, la demi-vie et d’extrapoler des données provenant d’une dose unique à une situation d’exposition chronique (Dybing et coll., 2002). Une telle approche ne permet cependant pas d’appréhender les cinétiques locales (tissulaires) ni d’effectuer des extrapolations interspécifiques car les modèles compartimentaux sont définis à partir des données plasmatiques et non à partir de considérations anatomiques, physiologiques ou métaboliques. Pour répondre au besoin d’extrapolation des modèles animaux à l’homme ou de l’in vitro à l’in vivo, ou encore pour connaître plus précisément l’exposition fœtale, des modèles plus complexes ont été élaborés, les modèles physiologiques ou PBPK (pour physiologically-based pharmacokinetic models). Ces modèles intègrent des données multiples parmi lesquelles les sites de distribution tissulaire, les enzymes impliquées dans le métabolisme, les cinétiques enzymatiques déterminées in vitro, les débits sanguins dans les principaux organes, les coefficients de partage tissu-sang, les constantes de liaison... (Leung, 1991, Clewell et Andersen, 1994).
). Une telle approche ne permet cependant pas d’appréhender les cinétiques locales (tissulaires) ni d’effectuer des extrapolations interspécifiques car les modèles compartimentaux sont définis à partir des données plasmatiques et non à partir de considérations anatomiques, physiologiques ou métaboliques. Pour répondre au besoin d’extrapolation des modèles animaux à l’homme ou de l’in vitro à l’in vivo, ou encore pour connaître plus précisément l’exposition fœtale, des modèles plus complexes ont été élaborés, les modèles physiologiques ou PBPK (pour physiologically-based pharmacokinetic models). Ces modèles intègrent des données multiples parmi lesquelles les sites de distribution tissulaire, les enzymes impliquées dans le métabolisme, les cinétiques enzymatiques déterminées in vitro, les débits sanguins dans les principaux organes, les coefficients de partage tissu-sang, les constantes de liaison... (Leung, 1991, Clewell et Andersen, 1994).Bibliographie
[1]ATSDR.;
(Agency for Toxic Substances and Disease Registry). Toxicological Profile for Polybrominated Diphenyl Ethers and Polybrominated Biphenyls. 2004 www.atsdr.cdc.gov/toxprofiles/tp68.html. 
[2] CLEWELL HJ, ANDERSEN ME. Physiologically-based pharmacokinetic modeling and bioactivation of xenobiotics.
Toxicol Ind Health. 1994;
10:1- 24
[3] DEKANT W. The role of biotransformation and bioactivation in toxicity.
EXS. 2009;
99:57- 86
[4] DYBING E, DOE J, GROTEN J, KLEINER J, O’BRIEN J, et coll. Hazard characterization of chemicals in food and diet: dose response, mechanisms and extrapolation issues.
Food Chem Tox. 2002;
40:237- 282
[5] GUENGERICH FP. Cytochrome P450 and chemical toxicology.
Chem Res Toxicol. 2008;
21:70- 83
[6] HAKK H, HUWE JK, LARSEN GL. Absorption, distribution, metabolism and excretion (ADME) study with 2,2’,4,4’,5,6’-hexabromodiphenyl ether (BDE-154) in male Sprague-Dawley rats.
Xenobiotica. 2009;
39:46- 56
[7] HATAGIMA A. Genetic polymorphisms and metabolism of endocrine disruptorsin cancer susceptibility.
Cad Saúde Pública. 2002;
18:357- 377
[8] HOLLOWAY AC, ANGER DA, CRANKSHAW DJ, WU M, FOSTER WG. Atrazine-induced changes in aromatase activity in estrogen sensitive target tissues.
J Appl Toxicol. 2008;
28:260- 270
[9] IKEZUKI Y, TSUTSUMI O, TAKAI Y, KAMEI Y, TAKETANI Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure.
Hum Reprod. 2002;
17:2839- 2841
[10] JANA S, PALIWAL J. Molecular mechanisms of cytochrome P450 induction: potential for drug-drug interactions.
Curr Drug Metab. 2007;
9:70- 76
[11] KESTER MHA, BULDUK S, VAN TOOR H, TIBBOEL D, MEINL W, et coll. Potent inhibition of estrogen sulfotransferase by hydroxylated metabolites of polyhalogenated aromatic hydrocarbons reveals alternative mechanism for estrogenic activity of endocrine disrupters.
J Clin Endocrinol Metab. 2002;
87:1142- 1150
[12] KITAMURA S, SUGIHARA K, SANOH S, FUJIMOTO N, OHTA S. Metabolic activation of proestrogens in the environment by cytochrome P450 system.
J Health Sci. 2008;
54:343- 355
[13] LEHMAN-MCKEEMAN LD. Absorption, distribution and excretion of toxicants. .
In: Casarett and Doull’s Toxicology – The basic science of poisons. In: Ed CD Klaassen, editors.
McGraw-Hill;
New York:2008;
131160
[14] LEUNG HW. Development and utilization of physiologically-based pharmacokinetic models for toxicological applications.
J Toxicol Environ Health. 1991;
32:247- 267
[15]OECD.OECD guideline for the testing of chemicals. Section 4: health effects. Test No 417: Toxicokinetics. 2010;
(http://www.oecd-ilibrary.org/environment/test-no-417- toxicokinetics_9789264070882-en).
[16] RIU A, CRAVEDI JP, DEBRAUWER L , GARCIA A, CANLET C, JOUANIN I, ZALKO D. Disposition and metabolic profiling of 14C-decabromodiphenylether in pregnant Wistar rats.
Environ Int. 2008;
34:318- 329
[17] SANDERS JM, LEBETKIN EH, CHEN LJ, BURKA LT. Disposition of 2,2’,4,4’,5,5’-hexabromodiphenyl ether (BDE153) and its interaction with other polybrominated diphenyl ethers (PBDEs) in rodents.
Xenobiotica. 2006;
36:824- 837
[18] SIMPSON ER, CLYNE C, RUBIN G, BOON WC, ROBERTSON K, BRITT K, SPEED C, JONES M. Aromatase – A brief overview.
Annu Rev Physiol. 2002;
64:93- 127
[19] SHIPKOVA M, WIELAND E. Glucuronidation in therapeutic drug monitoring.
Clinica Chimica Acta. 2005;
358:2- 23
[20] TOLSON AH, WANG H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR.
Adv Drug Deliv Rev. 2010, doi:10.1016/j.addr.2010.08.006;
[21] VANDENBERG LN, HAUSER R, MARCUS M, OLEA N, WELSHONS WV. Human exposure to bisphenol A (BPA).
Reprod Toxicol. 2007;
24:139- 177
[22] WALSH CT. Toxicokinetics: oral exposure and absorption of toxicants.
In: Comprehensive toxicology. In: BOND J (ed). Vol I General principles, editors.
Elsevier Science;
New York:1997;
5161
[23] WARING RH, AYERS S, GESCHER AJ, GLATT HR, MEINL W, et coll. Phytœstrogens and xenœstrogens: the contribution of diet and environment to endocrine disruption.
J Steroid Biochem Mol Biol. 2008;
108:213- 220
→ Aller vers SYNTHESE