III. Toxicologie
2013
21-
Mécanismes d’action des pesticides en cancérogenèse
Au cours des dernières décennies, de nombreux progrès ont été réalisés dans la compréhension de l’étiologie des cancers et des mécanismes fondamentaux de la cancérogenèse, suggérant la possibilité de causes génétiques et/ou environnementales. Le terme « cancérogène » se réfère à une classe de molécules (seules ou en association) qui, par elles même ou après métabolisation, provoquent l’apparition d’une tumeur ou augmentent son incidence et/ou sa malignité. Un composé sera considéré comme cancérogène si son administration aboutit, dans des conditions bien précises, au développement de tumeurs malignes au niveau d’un ou plusieurs tissus (Gomes-Carneiro et coll., 1997

; Huff, 1999
). Ainsi, la part attribuable de l’amiante dans la survenue de mésothéliomes est estimée à 80 % (McDonald et McDonald, 1996
; Anonyme, 1997
) et les radiations UV sont clairement le facteur environnemental prédominant dans celle du mélanome (Gandini et coll., 2011
). Cependant, les facteurs génétiques jouent également un rôle important dans la survenue de certains cancers (Xue et coll., 2012
; Xu et coll., 2012
) et certains polymorphismes génétiques peuvent être responsables d’une susceptibilité accrue aux effets cancérogènes de facteurs environnementaux (Hutter et coll., 2012
; Lesseur et coll., 2012
; Liu et coll., 2012
). Une séparation trop tranchée entre mécanismes génétiques et environnementaux semble réductrice puisque les polymorphismes génétiques et certaines modifications épigénétiques pourraient expliquer partiellement la susceptibilité individuelle aux effets toxiques de certains polluants environnementaux.
L’objectif de ce chapitre est d’analyser si les effets cancérogènes des pesticides, identifiés ou suspectés au travers des données épidémiologiques, peuvent être argumentés par leurs mécanismes d’action cellulaire et moléculaire.
Le nombre considérable de produits phytopharmaceutiques autorisés (auxquels s’ajoutent ceux non inscrits à l’Annexe de la Directive Européenne ou déjà interdits), qui doivent être pris en compte dans une telle analyse rétrospective, impose des critères de sélection.
La sélection des composés a été guidée par les critères suivants :
• les tonnages annuels en France (données UIPP et rapport Anses 2010
1
http://www.observatoire‑pesticides.fr/upload/bibliotheque/171959218396043870616875052847/exposition_population_generale_pesticides_2010_vdef.pdf" [lilen obsolète]
) ;
• le classement par le Circ (Centre international de recherche sur le cancer) et l’EPA (
Environmental Protection Agency) en termes de cancérogénicité (
annexe 4);
• les données épidémiologiques issues de la cohorte prospective AHS (
Agricultural Health Study) mise en place par le NCI (
National Cancer Institute) dès l993 (Weichenthal et coll., 2010
)
2
;
• l’analyse d’autres études épidémiologiques ayant fait l’objet de cette expertise.
Les composés sélectionnés ont ensuite été soumis à une recherche bibliographique par mots clés, ciblant les principaux mécanismes susceptibles d’intervenir dans la cancérogenèse : la génotoxicité, la régulation des processus de survie cellulaire (voie de signalisation de l’apoptose, la nécrose, l’autophagie), l’implication de récepteurs nucléaires et/ou hormonaux (ER, AR, PXR, CAR…), l’induction des systèmes de biotransformation (cytochromes P450 en particulier), la génération de stress oxydant…
Une attention particulière a été portée à :
• la qualité scientifique et méthodologique des données des publications examinées (Impact Factor > 1,5) ;
• leur actualité au regard des évolutions réglementaires et technologiques ;
• la pertinence des modèles in vivo (transposablité animal-homme) et in vitro (lignées versus primo-cultures, organospécificité, bioactivation), ainsi que des tests toxicologiques ou des biomarqueurs de cancérogenèse/génotoxicité, retenus dans ces études expérimentales ;
• la représentativité des expositions au regard des scénarii pouvant intervenir chez l’Homme (niveaux de doses, durée et chronicité ou non, modalités d’administration, présence de véhicules ou de mélanges).
Dans la mesure du possible, les Valeurs Toxicologiques de Référence (VTR) des molécules ont également été prises en considération dans cette analyse : doses et effets critiques retenus dans leur élaboration, effets seuil versus sans seuil…
Processus de cancérogenèse
La cancérogenèse
3
On parle également de processus tumoral ou néoplasique, caractérisé par une multiplication de cellules anormales (cellules tumorales, néoplasiques) qui se divisent par mitose pour générer une tumeur maligne (tumorigenèse).
(ou carcinogenèse, ou oncogenèse) constitue un processus multifactoriel au cours duquel des modifications génotypiques et phénotypiques concourent à l’apparition d’un clone de cellules transformées à avantage sélectif de croissance. Les xénobiotiques pro-carcinogènes et/ou leurs métabolites actifs affectent l’expression de gènes impliqués dans la régulation du cycle cellulaire, de la réparation de l’ADN ou de l’apoptose. Qu’elles soient génotoxiques ou non, ces molécules peuvent interférer avec la transduction des signaux et aboutir
in fine à une hypermutabilité, une instabilité génomique, une perte du contrôle de la prolifération cellulaire et une résistance à l’apoptose.
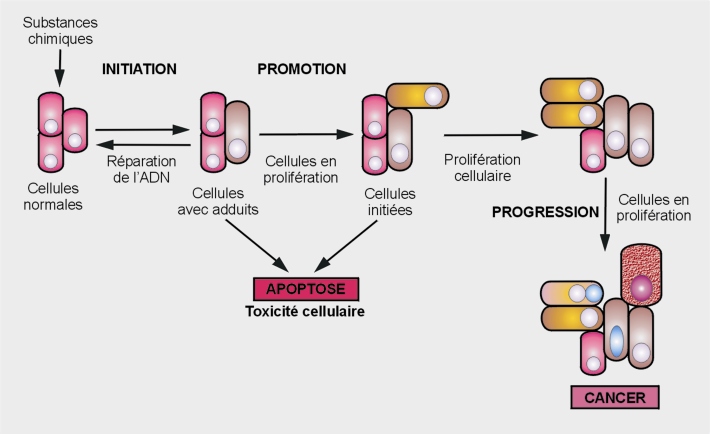
Étapes de la tumorigenèse
Les trois étapes du développement néoplasique rendent compte de bouleversements structuraux conséquents dus à des atteintes du génome et/ou des perturbations de signaux cellulaires et moléculaires (figure 21.1
).
Initiation
La phase d’initiation tumorale est causée par des modifications de l’ADN irréversibles qui prédisposent des cellules normales à l’acquisition de la capacité de prolifération non contrôlée et indéfinie (immortalisation) et à une évolution maligne. Ces cellules ne sont pas, à proprement parler, néoplasiques mais elles ont subi les altérations génotypiques nécessaires pour le devenir. Contrairement au génotype, ces cellules initiées présentent le même phénotype cellulaire que les cellules normales.
Après exposition à un cancérogène, les dommages occasionnés à l’ADN peuvent être réparés par des mécanismes enzymatiques spécifiques après arrêt du cycle cellulaire. Si la réparation ne peut avoir lieu (nombre de mutations ou gravité trop importants), la cellule s’engage alors dans un processus de mort cellulaire programmée (apoptose). La division cellulaire est essentielle dans cette étape car si elle se produit avant la mise en œuvre des systèmes de réparation à l’ADN, les dommages deviennent permanents et irréversibles (phase de fixation de la mutation dans le génome). L’initiation est un phénomène rapide et irréversible qui assure une descendance cellulaire parfaitement identique à la cellule mutée (Heidelberger, 1977
; Farber, 1984
; Richardson et coll., 1986
; Frowein, 2000
). À cette étape, la cellule initiée peut rester quiescente pendant une période indéfinie (allant du jour à l’année), ou alors se diviser de manière autonome et clonale. Cette division reste modérée et contrôlée.
C’est un processus additif car le développement néoplasique dépend de la dose en composé cancérogène. Ainsi, une augmentation de cette dose augmente l’incidence et la multiplicité des foyers néoplasiques résultants et diminue la période de latence de sa survenue.
Promotion
La phase de promotion tumorale se caractérise par une forte instabilité génomique et correspond à la prolifération (multiplication anormale) clonale des cellules initiées.
Les promoteurs tumoraux n’interagissent pas directement avec l’ADN et peuvent déclencher des effets biologiques sans avoir été activés métaboliquement (Yuspa et coll., 1983
; Williams, 2001
). Ils augmentent la prolifération cellulaire dans les tissus cibles (par levée du blocage du cycle cellulaire en phase quiescente ou G0), contribuent à la « fixation » des mutations, potentialisent des modifications de l’expression génique, et perturbent le contrôle de l’apoptose, processus de sauvegarde. Ils peuvent également altérer de façon indirecte l’ADN par oxydation (génération de stress oxydant).
Les promoteurs tumoraux doivent être présents pendant une très longue période pour agir et leur efficacité dépend de leur concentration dans le tissu cible (Butterworth et coll., 1992
). Dans les études menées sur les composés cancérogènes sur de longues périodes avec des doses élevées, pratiquement tous les agents promoteurs de tumeurs peuvent induire un processus de néoplasie sans initiation (Pitot et Dragan, 1991
). Par exemple, l’exposition au phénobarbital ou à l’arsenic aboutit au développement de lésions néoplasiques sans application préalable à des agents initiateurs.
Très généralement, la promotion est une étape réversible : après disparition du promoteur, la prolifération cellulaire peut être atténuée voire même stoppée.
Progression
Les lésions survenant au cours des phases d’initiation et de promotion sont définies comme pré-néoplasiques. Leur transformation en lésions malignes est associée à l’une des dernières étapes de la cancérogenèse, appelée progression. Au cours de cette étape, un phénotype néoplasique est acquis via des mécanismes génétiques et épigénétiques. La prolifération cellulaire devient indépendante de la présence du stimulus. Cette phase se caractérise par l’irréversibilité, l’instabilité génétique, une croissance cellulaire très rapide, l’invasion et la capacité des cellules à métastaser, ces caractéristiques étant associées à des modifications biochimiques, métaboliques et morphologiques des cellules. L’angiogenèse est un processus essentiel à la progression néoplasique puisqu’il précède le développement des caractéristiques qui contribuent à la malignité. De nouvelles modifications inhérentes à la néovascularisation cellulaire apparaissent (fabrication de nouveaux vaisseaux) créant une interface entre la tumeur et les tissus de voisinage : le stroma. À ce stade, la dimension de la tumeur atteint quelques millimètres. L’angiogenèse se développe et les cellules cancéreuses élaborent une architecture plus complexe. Cette phase dite phase de stroma-réaction précède les métastases.
Métastases et transition épithélio-mésenchymateuse
Lors de la progression tumorale, l’acquisition de propriétés métastatiques constitue une étape tardive induite en réponse à la détérioration des conditions environnementales de la tumeur. Le processus métastatique correspond à la dissémination de cellules à partir d’une tumeur primaire vers des organes à distance. Cette cascade d’évènements définit l’agressivité d’une tumeur, fait appel à l’acquisition des propriétés de mobilité cellulaire et nécessite la mise en jeu d’un processus fondamental : la transition épithélio-mésenchymateuse (TEM). La TEM est un processus physiologique indispensable lors de l’embryogenèse (type 1) et dans des situations de régénération ou réparation comme la cicatrisation (type 2), mais elle intervient également dans la formation des métastases (type 3) (Shook et Keller, 2003
). La TEM regroupe 4 grands types de modifications : le remodelage du cytosquelette, une altération (voire la dissociation totale) des structures de contacts entre les cellules, notamment les jonctions adhérentes qui implique la E-cadhérine, molécule d’adhérence assurant la cohésion cellule-cellule de l’épithélium, une modification des interactions avec la matrice extra-cellulaire (MEC) sous-jacente et une dégradation de celle-ci par des protéases (Lauffenburger et Horwitz, 1996
; Boyer et coll., 2000
; Thiery et Sleeman, 2006
).
La progression tumorale implique une succession d’évènements complexes au cours desquels les interactions entre cellule-cellule et cellule-MEC environnante vont être modifiées. Certaines cellules peuvent se dissocier du foyer tumoral primaire pour acquérir de nouvelles propriétés. Ces cellules adoptent un phénotype distinct (métastable) coïncidant avec la perte du caractère épithélial (Thiery, 2003
; Lee et coll., 2006
).
La répression génique de la E-cadhérine (facteur clé du processus de TEM) entraîne la dissociation du complexe E-cadhérine/β-caténine présent au niveau des jonctions adhérentes (Kalluri et Neilson, 2003
; Huber et coll., 2005
). En parallèle, différentes perturbations intra-cellulaires se produisent telles que la perte de la polarisation de la cellule, la modulation de l’expression des molécules responsables de l’adhésion cellulaire (perte de marqueurs épithéliaux comme la E-Cadhérine, la cytokératine et l’occludine), l’acquisition de marqueurs de cellules mésenchymateuses tels que la N-Cadhérine, la vimentine, la ténascine C et un réarrangement des filaments d’actine en fibres de stress, phénomène conférant aux cellules leur plasticité et capacité migratoire (Savagner, 2001
; Christofori, 2006
). Il est à noter que de nombreux gènes impliqués dans le développement sont ré-exprimés dans les cellules invasives (Snail, Slug, Twist ou Notch).
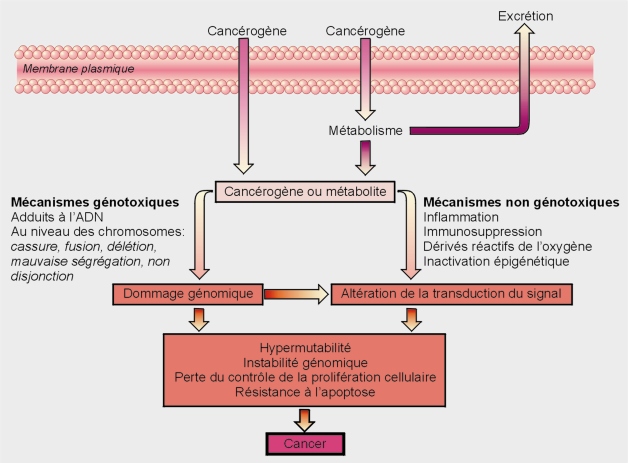
Classement des substances par mode d’action principal
Les polluants physico-chimiques sont souvent classés traditionnellement selon deux modes d’action principaux, en génotoxiques (agents initiateurs de cancers) ou non génotoxiques (agents promoteurs).
Substances cancérogènes génotoxiques
Une substance cancérogène génotoxique est une substance capable de provoquer l’apparition de tumeurs (bénignes/malignes) en altérant la transmission fidèle du génome d’une génération de cellules à l’autre (altération du matériel génétique). Elle peut être mutagène, clastogène et/ou aneugène.
Une substance mutagène provoque des mutations au niveau du matériel génétique (ADN). Elle est capable, par elle-même ou par ses métabolites, de provoquer une tumeur en induisant et en augmentant la fréquence des mutations. Elle peut induire des mutations sur des portions de gènes spécifiques critiques, le plus souvent des proto-oncogènes, des gènes suppresseurs de tumeur et/ou des gènes de réparation de l’ADN. Elle ne peut être mutagène qu’à condition d’altérer la structure du gène de façon permanente et transmissible aux cellules filles par mitoses successives. Les systèmes de défense permettant à la cellule de se protéger de l’induction des mutations permanentes ou de les réparer sont débordés et ne peuvent plus restaurer l’intégrité du matériel génétique ou empêcher la transmission de mutations. Les mutations peuvent impliquer un gène unique (mutation ponctuelle), un ensemble de gènes (mutation chromosomique), un ou plusieurs chromosomes entiers (mutation génomique).
Une substance clastogène est capable de provoquer l’apparition d’une tumeur par des modifications structurales des chromosomes (cassure du matériel génétique). Une substance aneugène est capable de provoquer une tumeur en altérant la répartition des chromosomes modifiant ainsi le nombre de chromosomes (aneuploïdie). Le mécanisme résulte soit de l’altération des protéines constitutives de l’appareil mitotique, soit de la formation d’adduits encombrants (bulky adducts) empêchant ainsi la bonne ségrégation des chromosomes.
Substances cancérogènes non génotoxiques
Une substance cancérogène non génotoxique est une substance capable de provoquer l’apparition d’un foyer néoplasique sans action directe sur le matériel génétique (sans altération de l’ADN ou de la structure et du nombre de chromosomes) via une stimulation indirecte de réponses hyperplasiques. Ces composés peuvent avoir de multiples mécanismes conduisant au cancer. Le mode d’action non génotoxique inclut des effets qui n’impliquent pas des altérations de l’ADN, mais influencent l’expression génique, la communication entre cellules ou d’autres facteurs du processus de cancérogenèse. Les effets cancérogènes épigénétiques (méthylation de l’ADN, remodelage de la chromatine) reposent sur ce mécanisme (promoteurs tumoraux).
De cette multitude de modes d’action découle la très grande difficulté de caractériser ces composés comme étant cancérogènes. De plus, ces molécules peuvent avoir différents modes d’action qui sont pour la plupart, tissus et espèces spécifiques.
Pour ces molécules, la survenue de cancer nécessite l’altération de nombreuses voies de signalisation. Parmi ces dernières, on peut citer celles qui conduisent à la promotion tumorale, aux modifications endocrines, à la suppression du système immunitaire, à la toxicité tissu-spécifique (apoptose, nécrose) et aux réponses inflammatoires.
Les deux grands types de processus de cancérogenèse sont présentés dans la figure 21.2
.
Mécanismes cellulaires et moléculaires impliqués
Mutations des gènes
Les phénomènes de génotoxicité peuvent concerner des gènes clés du développement d’un clone de cellules tumorales. Ces gènes, classés en deux catégories, contrôlent l’ensemble des réactions métaboliques impliquées dans la progression du cycle cellulaire, soit de manière positive avec gain de fonction (oncogènes, proto-oncogènes), soit de manière négative avec perte de fonction (suppresseurs tumoraux).
Si la structure du gène est altérée de façon permanente et transmissible aux cellules filles, les systèmes de défense permettant à la cellule de se protéger de l’induction des mutations permanentes sont débordés et ne peuvent plus restaurer l’intégrité du matériel génétique ou empêcher la transmission de mutations.
Les proto-oncogènes codent pour des oncoprotéines ayant un équivalent dans le génome d’un virus. Ces oncoprotéines sont impliquées dans la régulation positive de la prolifération cellulaire à différents niveaux de la transmission des signaux contrôlant notamment la prolifération cellulaire (facteurs de croissance et leurs récepteurs, protéines G, facteurs de transcription… ras, N-myc, c-jun, c-fos, c-erbB, c-myc, TAll1, Abl, PML). L’implication des proto-oncogènes dans les processus de cancérogenèse nécessite une activation entraînant leur surexpression (gain de fonction) sur un mode dominant positif (la mutation d’un seul allèle suffit). Leur activation survient à toutes les étapes de la cancérogenèse par différents mécanismes (mutations somatiques ponctuelles, amplification génique, réarrangement génique). L’accumulation des mutations au niveau de différents oncogènes potentialise leurs effets transformants et aboutit à l’apparition d’une tumeur (coopération oncogénique). Dans de nombreux cancers, une ou plusieurs altérations géniques supplémentaires sont nécessaires, en particulier l’inactivation de gènes suppresseur de tumeurs.
Les gènes suppresseurs de tumeur (anti-oncogènes) codent pour des protéines associées à l’arrêt du cycle cellulaire, à l’apoptose et à la réparation des lésions de l’ADN. Ils sont rendus inactifs par mutation dans les régions codantes, par inhibition de la transcription, par délétion ou aneugenèse. Ils interviennent dans la cancérogenèse par perte de fonctions généralement sur un mode récessif (nécessite l’inactivation des deux allèles). Cette catégorie comprend les gènes codant pour p53 (50 % des cancers humains), p19ARF, Rb1 (rétinoblastome), WT1, NF1, APC (colon), BCRA1, BCRA2 (sein)… Il existe des cancers qui sont dus à l’absence héréditaire de plusieurs anti-oncogènes, expliquant ainsi les prédispositions familiales à certains cancers.
Outre les gènes suppresseurs de tumeur, la cellule peut réagir par la mise en œuvre des systèmes de réparation de l’ADN, gènes « care taker ». Six grands systèmes de réparation existent au sein des cellules vivantes : des mécanismes ad hoc, spécifiques d’un type de lésion donnée (photo-lyase, méthyltransférases), et cinq systèmes généralistes, chacun capables de réparer un ensemble de lésions diverses (réparation par excision de nucléotides, réparation des mésappariements, réparation par religation non homologue et réparation par recombinaison homologue).
Activation métabolique des cancérogènes
Qu’il soit génotoxique ou non-génotoxique, un composé peut présenter une activité cancérogène directe (de part ses propriétés intrinsèques) ou après activation par les voies métaboliques (composés cancérogènes indirects ou pro-cancérogènes). Ces activations métaboliques sont en général dépendantes des enzymes de phase I et notamment des cytochromes P450 (CYPs). L’introduction par ces derniers d’un groupe polaire réactif convertit le composé initial en un produit électrophile capable de former des adduits à l’ADN. Les enzymes de phase II peuvent néanmoins transformer ces métabolites réactifs en composés inertes facilement éliminables. Malgré la nécessité de biotransformation pour éliminer les molécules chimiques de l’organisme, celle-ci peut avoir des effets adverses très délétères concourant à l’activation de certaines molécules pro-cancérogènes. Ce processus est essentiellement retrouvé dans le foie, organe majeur de la détoxication réalisant la majorité des réactions de biotransformation. Dans ce sens, cet organe constitue un organe effecteur mais également cible de la toxicité des composés réactifs éventuellement générés par les enzymes du métabolisme des xénobiotiques (EMX) de phase I.
Parallèlement à ce processus de biotransformation, des phénomènes de peroxydation peuvent se produire, engendrant la création de dérivés réactifs de l’oxygène (DRO). Ils provoquent des atteintes à l’ADN, l’ARN et aux protéines par l’intermédiaire de réactions chimiques telles que l’oxydation, la nitration/nitrosation, et l’halogénation. Ces phénomènes entraînent un risque potentiel d’augmentation des mutations et des altérations dans les fonctions de protéines et d’enzymes majeures. En outre, les DRO produits peuvent influer sur différents processus vitaux (maintien du statut redox de la cellule, métabolisme, transduction du signal régulant l’oxydation ou la réparation de l’ADN…).
La spécificité des systèmes d’activation des différents tissus, régule le développement néoplasique et dépend du polymorphisme génétique qui régule l’expression et la distribution des EMX. Par exemple, une personne qui a une grande quantité d’enzymes de phase I et de faibles quantités d’enzyme de phase II a plus de probabilité de synthétiser des composés intermédiaires réactifs, susceptibles d’entraîner des dommages à l’ADN. Il en résulte donc un terrain de « prédisposition » à la survenue de tumeurs inhérentes aux processus de bioactivation.
Stress oxydant
Le développement de tumeurs par des molécules non génotoxiques est aggravé par des facteurs tels que la génération de stress oxydant, le démantèlement des jonctions intercellulaires et les propriétés acquises des cellules.
Le stress oxydant se définit par un déséquilibre entre la production de dérivés réactifs de l’oxygène (DRO) et les capacités anti-oxydantes de la cellule. Ces dérivés réactifs de courte durée de vie comprennent : le radical hydroxyle (OH•), l’anion superoxyde (O2•-), le peroxyde d’hydrogène (H2O2) et l’oxygène singulet (1O2). En raison de leur extrême réactivité les radicaux OH• sont les plus toxiques. Puissants agents oxydants, ils s’attaquent à la plupart des molécules cellulaires : ADN, protéines, lipides (membranes), acides aminés, sucres et métaux. Les radicaux O2•- semblent moins réactifs. Toutefois, ces derniers demeurent des espèces potentiellement toxiques via leurs réactions avec le peroxyde d’hydrogène (H2O2) et monoxyde d’azote (NO), générant respectivement des radicaux OH• et des peroxynitrites. Ces deux espèces sont particulièrement réactives vis-à-vis des cibles biologiques.
Une surproduction de DRO ou un déficit en systèmes antioxydants (superoxyde dismutase, glutathion, glutathion peroxydase, catalase…) peut aboutir à un déséquilibre ou « stress oxydant » qui peut conduire à une mort cellulaire par apoptose ou nécrose. En effet, les DRO peuvent induire une dépolarisation mitochondriale aboutissant à la libération du cytochrome C, responsable de l’activation en cascade des caspases, effectrices de l’apoptose. De plus, lorsque leur production dépasse les capacités de défense de la cellule, ils peuvent exercer un effet délétère direct par oxydation de résidus protéiques, lipidiques, nucléotidiques, en s’attaquant aux constituants cellulaires vitaux… De ce fait, le stress oxydant est impliqué dans l’étiologie de nombreuses maladies (néoplasiques, neurodégénératives, inflammatoires, cardiovasculaires…) ainsi que le vieillissement.
Cependant, hormis ces effets délétères, la production contrôlée de DRO apparaît de plus en plus comme un mécanisme essentiel participant au maintien de l’homéostasie cellulaire, constituant un véritable système de signalisation cellulaire. Ces dérivés sont en effet utilisés comme second messagers contrôlant étroitement la survie/mort, la prolifération ou la différenciation cellulaire, par l’intermédiaire de nombreux facteurs transcriptionnels (HSF1, NF-kappaB, P53), et de kinases (PI(3)K, AKT, MAPK, ERK, JNK et P38 kinase…). Leur production intracellulaire sous l’influence de facteurs exogènes ou endogènes (xénobiotiques, irradiation UV ou RX, inflammation, cytokines…), intervient à plusieurs niveaux : transmission du signal, régulation transcriptionnelle et post-transcriptionnelle…
Liaison aux récepteurs nucléaires
Certains cancérogènes non-génotoxiques sont des perturbateurs endocriniens car ils se lient directement aux récepteurs ER, à celui de la progestérone, au récepteur AhR ou encore au récepteur à l’hormone thyroïdienne. Une déstabilisation de la balance dans les signalisations impliquées dans l’homéostasie de ces hormones, couplés à la cytotoxicité, l’hyperplasie et l’inhibition des jonctions communicantes peut constituer des évènements clés dans l’induction d’effets promoteurs du développement de cancers.
Apoptose/nécrose
La mort cellulaire induite par les agents cytotoxiques est le plus souvent de type apoptotique, dans les cellules tumorales comme dans les cellules normales. L’apoptose est un processus physiologique, génétiquement programmé et déterminant dans la morphogenèse, le vieillissement et l’élimination des cellules potentiellement dangereuses. Certains stimuli pro-apoptotiques endogènes ou exogènes (stress oxydant, xénobiotiques, UV…) déclenchent différentes voies de signalisation aboutissant à l’activation des caspases puis à la mort de la cellule. La régulation de ce processus est sous contrôle de protéines, dont les mieux caractérisées sont celles de la famille Bcl-2. Un déséquilibre entre les formes pro et anti-apoptotiques orientera la cellule vers sa survie ou sa mort. Il est bien établi que les dérèglements de l’apoptose sont primordiaux dans la formation de tumeurs, puisque les cellules cancéreuses ont perdu leur capacité à se suicider par ce processus actif. Ainsi, les cellules cancéreuses présentent fréquemment une altération des voies d’induction ou d’inhibition de l’apoptose et plusieurs formes de cancers sont liées à une suppression atypique des protéines anti-apoptotiques telles que Bcl2, Bcl-xL, Mcl-1…
Cas particuliers des hépatocarcinomes
Les hépatocarcinomes (HCC) constituent le 8e cancer dans le monde. S’il survient le plus souvent de manière consécutive à une cirrhose, plusieurs études relatent une probable incidence des contaminants environnementaux sur la survenue des HCC. Ainsi, de nombreuses études menées in vivo chez l’animal ou in vitro suggèrent que certains pesticides présentent des potentialités pro-cancérogènes. Ils peuvent être des promoteurs tumoraux, influer sur la potentialisation ou la survenue d’un processus métastasique ou encore favoriser des états pré-néoplasiques ou des facteurs de prédisposition aux hépatocarcinomes (état fibrotique). La pathogenèse des hépatocarcinomes est multifactorielle et comprend des composantes environnementales, infectieuses, nutritionnelles, métaboliques et endocrines.
Le foie est le seul organe de l’organisme à être doué de régénération. La division des hépatocytes est rare dans le foie sain car ces cellules sont bloquées en phase G0 du cycle cellulaire (Fausto et Webber, 1993
; Michalopoulos et DeFrances, 1997
). Toutefois, après une atteinte hépatique ou une hépatectomie, les hépatocytes reçoivent de multiples signaux aboutissant à leur prolifération et donc à la reconstruction du tissu hépatique. La régulation physiologique de la balance entre la prolifération et l’apoptose hépatocytaire est essentielle pour l’homéostasie du foie. Ainsi, tout dérèglement peut aboutir à une hyperplasie et au développement de tumeurs hépatiques (Klaunig et coll., 2000
). Le contrôle de cette balance est notamment opéré par le HGF (
Hepatocyte Growth Factor), responsable de la prolifération des hépatocytes et le TGF-β (
Transforming Growth Factor β) conduisant à la mort de ces cellules par apoptose.
Après nécrose hépatique, les hépatocytes quiescents prolifèrent, permettant ainsi le renouvellement cellulaire. Les cycles répétitifs de nécrose/régénération constituent des phénomènes qui facilitent l’acquisition d’altérations génomiques et donc la survenue des hépatocarcinomes. Il est bien établi que la régénération incontrôlée constitue un facteur important de l’hépatocarcinogenèse (Röcken et Carl-McGrath, 2001
).
Les composés hépatocarcinogènes non-génotoxiques (tels que le phénobarbital, le TCPOBOP, la dioxine, le clofibrate et des pesticides tels que la dieldrine) stimulent la promotion tumorale en altérant les processus de prolifération, d’apoptose et de différenciation cellulaire (Oliver et Roberts, 2002
). Différentes études ont suggéré que l’apoptose hépatocytaire est essentielle dans les trois étapes de l’hépatocarcinogenèse : l’initiation, la promotion et la progression par expansion clonale (Grasl-Kraupp et coll., 1994
). Le développement des hépatocarcinomes est probablement dû, au moins en partie, à la surexpression de protéines anti-apoptotiques, telles que les IAPs (et plus particulièrement la survivine) et Bcl-xL (Takehara et coll., 2001
; Ikeguchi et coll., 2002
). De plus, les HCC adoptent différentes stratégies pour échapper à l’apoptose initiée par les cellules immunitaires. Ainsi, les hépatocytes tumoraux n’expriment plus le Fas et ne sont donc plus sensibles à l’apoptose induite par le FasL (
Fas Ligand) sécrété. De plus, ces cellules surexpriment le FasL, processus leur permettant de tuer les lymphocytes qui leurs seraient fatals. Elles présentent également une perte d’expression ou une mutation du récepteur au TGF-β, phénomène favorisant leur survie et leur prolifération (Kiso et coll., 1994
; Moller et coll., 1994
).
Pesticides et évaluations des risques cancérogènes
Évaluation des risques cancérogènes au niveau réglementaire
Études de génotoxicité
Les études ont pour but d’identifier la nature des altérations du matériel génétique : pouvoir mutagène direct vis-à-vis de l’ADN, aberrations de la structure chromosomique (clastogenèse) ou de leur nombre (potentiel aneugène). Elles reposent sur tout un éventail de tests réglementaires pratiqués in vitro et/ou in vivo (Directives 91/414/CEE et 67/548/CEE, conformes aux lignes directrices de l’OCDE) : mutations géniques inverses sur bactéries (OCDE 471, E. coli, S. typhimurium) ou sur cellules eucaryotes (OCDE 476, mouse lymphoma TK +/- assay), aberrations chromosomiques et micronoyaux (perte de chromosomes entiers ou de fragments non intégrés au noyau durant la mitose) qui seront détectés sur des cellules de mammifère in vitro (OCDE 473, lymphocytes, lignée CHO15) ou in vivo (OCDE 474-475, cellules de moelle osseuse, érythrocytes ou lymphocytes périphériques). D’autres tests complémentaires visant à détecter des altérations primaires de l’ADN seront si nécessaire mis en place : mesure de synthèse non programmée de l’ADN hépatocytaire (ou UDS, mettant en évidence la capacité de réparation de l’ADN après action d’un toxique, OCDE 486), détection d’échange de chromatides sœurs sur cellules de mammifères (SCE, OCDE 479), mesures des adduits à l’ADN in vitro ou in vivo… Ces études conditionnent l’élaboration des valeurs toxicologiques de référence (VTR) puisqu’un produit génotoxique est « historiquement » et réglementairement considéré à ce jour comme étant dénué de seuil d’action en fonction de la dose. En effet, pour un tel produit et quelle que soit sa dose, il existe théoriquement un risque qu’une seule molécule, puisse affecter le matériel génétique (notamment par mutation) et initier une cascade d’événements déclenchant une cancérogenèse. Cette distinction est cependant débattue pour les composés aneugènes ou clastogènes, pour lesquels un mécanisme d’action à seuil est quelquefois retenu.
Effets cancérogènes
Les effets sont identifiés par la présence de tumeurs malignes résultant d’une prolifération excessive et non coordonnée de cellules initialement saines, dans le tissu d’origine. Plusieurs nomenclatures de classification et critères de diagnostic tumoral complémentaires ont été élaborées pour faciliter leur harmonisation (Circ,
Society of Toxicology Pathology, Registry of Industrial Toxicology Animal Data, OMS…). Pour les mettre en évidence, la Directive 91/414/CEE préconise la mise en place d’études (ou d’une étude combinée) de toxicité chronique et de cancérogenèse sur deux espèces (vie entière : 2 ans chez le rat, 18 mois chez la souris, OCDE 451-453
4
). Une attention très particulière doit être portée sur les « témoins historiques », l’éventuel cumul des incidences tumorales et les critères de qualité des études expérimentales (cotation de Klimisch
5
La cotation des études en se basant sur l’approche de Klimisch et coll. (1997
) prend en compte la fiabilité (méthodes standardisées, Bonnes Pratiques de Laboratoire (BPL), détail de description technique de la publication), la pertinence, et l’utilité des données dans le cadre de l’évaluation du risque. Cette cotation varie de 1 à 4.
: BPL, protocoles validés et normalisés…).
Classification (cancérogénicité)
Les molécules cancérogènes chez l’Homme ont été classée par le Circ en différentes catégories selon les études réglementaires, mécanistiques et épidémiologiques : Groupe 1 : l’agent est cancérogène pour l’homme ; Groupe 2A : l’agent est probablement cancérogène pour l’homme ; Groupe 2B : l’agent est peut-être cancérogène pour l’homme ; Groupe 3 : l’agent est inclassable quant à sa cancérogénicité pour l’homme ; Groupe 4 : l’agent n’est probablement pas cancérogène pour l’homme
Une seconde classification, réalisée par l’EPA (US Environmental Protection Agency), regroupe les pesticides cancérogènes selon 5 catégories : Groupe A : l’agent est cancérogène pour l’Homme ; Groupe B1 : l’agent est probablement cancérogène pour l’Homme mais les « preuves épidémiologiques » sont limitées et il est impossible de conclure ; Groupe B2 : l’agent est probablement cancérogène pour l’Homme mais les preuves sont suffisantes chez l’animal mais pas chez l’Homme d’après les données épidémiologiques ; Groupe C : l’agent est peut être cancérogène chez l’Homme ; Groupe D : l’agent est inclassable quant à sa cancérogénicité pour l’Homme ; Groupe E : il y a des preuves comme quoi le pesticide n’est pas cancérogène chez l’Homme.
Estimation des risques cancérogènes des pesticides
L’estimation des risques cancérogènes nécessite, d’une part le calcul des niveaux d’exposition journaliers par voie cutanée, ingestion et inhalation, selon les populations concernées, aboutissant à une dose journalière d’exposition au produit phytosanitaire (DJE) et d’autre part, l’élaboration des valeurs toxicologiques de références (VTR) pour ce produit, qui sera spécifique de son mode d’exposition. La détermination de ces dernières sera effectuée à partir des données expérimentales et/ou épidémiologiques en règle générale, selon deux cas de figures.
Produits non-génotoxiques
Lorsque le caractère non-génotoxique de la molécule a été démontré au travers de la batterie de tests réglementaires ou de recherches plus fondamentales sur les mécanismes d’action, il est considéré que l’effet cancérogène ne survient que si une certaine dose est atteinte et dépasse les capacités de détoxication de l’organisme. Dans ce cas, une VTR, notamment la dose journalière admissible ou DJA (quantité de produit auquel un individu peut-être théoriquement être exposé toute sa vie sans effet sanitaire nuisible) prenant en compte l’existence d’un tel seuil d’action dose-dépendant est estimée. Celle-ci, exprimée en mg.kg-1.j-1 ou en mg.m-3 (inhalation) est, selon les données expérimentales disponibles, estimée en divisant la dose maximale observée sans manifestation d’effets cancérogène ou pré-cancérogènes avérés (NOAEL) et/ou la dose minimale entraînant ces mêmes effets (LOAEL), par différents facteurs d’incertitudes. Ces derniers rendent compte des éventuelles variabilités inter-espèces (facteur 10) et polymorphismes de réponse inter-individuelle chez l’homme (facteur 10), d’ordres toxicocinétique et toxicodynamique. Un facteur d’incertitude (uncertainty factor, UF) de 100 est donc généralement appliqué, mais il peut être renforcé, selon la qualité ou la fiabilité insuffisantes des données expérimentales (x 3-10), la sévérité de l’effet (x 3-10), la nature des doses critiques (NOAEL, LOAEL) ou des études retenues (chroniques à sub-chroniques) pour les estimer (facteur 3 et 10, respectivement). Soulignons que malgré l’application quasi-généralisée de cette démarche, les doses critiques estimées à partir des NOAEL/LOAEL sont de plus en plus remises en question du fait de leurs incertitudes (taille de l’échantillon, niveau de précision, intervalle de confiance, extrapolation des doses auxquelles l’effet cancérogène se manifeste…). L’approche BMR (Benchmark Response) lui est préférée, notamment par l’US EPA car elle permet de déterminer la dose critique en fonction du niveau de réponse toxicologique ou du pourcentage d’excès de cette réponse par rapport aux contrôles, permettant de minimiser les sources d’imprécisions expérimentales.
Au total, un indice de risque pour la population générale sera déterminé par le rapport DJE (voie alimentaire) ou CI (concentration inhalée)/VTR (spécifique de la voie d’exposition). Un risque sanitaire est supposé exister si ce rapport est supérieur à 1. Pour information et bien que cela n’entre pas dans le cadre de l’évaluation du risque cancérogène des pesticides, ajoutons qu’en ce qui concerne les opérateurs, il est procédé à la détermination d’un niveau acceptable d’exposition (AOEL), correspondant à la dose maximale de pesticides à laquelle l’agriculteur peut être quotidiennement soumis sans impact sanitaire. La VTR correspondante est construite à partir des doses critiques obtenues d’après les études toxicologiques inférieures ou égales à 90 j, divisées par les facteurs de sécurité précédemment évoqués.
Produits génotoxiques
Lorsque le produit est génotoxique et qu’il ne présente donc pas, par hypothèse, de seuil d’action dose-dépendant, l’estimation de la VTR est plus complexe et ne fait à ce jour pas l’objet d’un consensus scientifique international. Extraite des données toxicologiques expérimentales et/ou épidémiologiques, elle est généralement exprimée en Excès de Risque Unitaire (ERU). Le point délicat de son estimation est la sélection des modèles les plus pertinents d’interpolation des données de toxicologie expérimentales ayant servi à la détermination des doses critiques (NOAEL, AOEL, BMDL (Benchmark Dose (Lower Confidence Limit)), ainsi que d’extrapolation des données (relation dose-effet) vers les très faibles doses, jusqu’à un niveau de risque cancer considéré comme acceptable (10-6 pour la plupart des agences). Le modèle d’interpolation appliqué permet de retenir le point de départ (POD) le plus pertinent de la courbe dose-réponse, permettant une extrapolation vers les faibles doses. Estimée par extrapolation linéaire à l’origine à partir de ce POD, l’ERU (appelé aussi slope factor) constitue ainsi la VTR des produits génotoxiques, exprimée en [mg.kg-1.j-1]-1 (voie orale) ou [mg.m-3]-1 (voie respiratoire). Ces niveaux de doses ou de concentrations sont associés à différents niveaux de risque (10-4, 10-5, 10-6), correspondant à la probabilité supplémentaire par rapport à un sujet non exposé qu’un individu contracte un cancer s’il est soumis toute sa vie à une unité de dose du produit. Un excès de risque individuel ou ERI (DJE ou CI x ERU x temps d’exposition) sera estimé : il sera considéré comme inacceptable en population générale s’il est supérieur à 10-4, qualifié de non-significatif s’il est inférieur à 10-6.
L’établissement des VTR des cancérogènes sans effet seuil est, d’autant plus aléatoire qu’il nécessite d’extrapoler à l’Homme les données expérimentales, vers les très faibles doses. Enfin, la classification même entre molécules cancérogènes génotoxiques (directement ou via leur métabolisme) et non-génotoxiques (dérégulation des voies signalétiques…) reste difficile à faire. Il en résulte qu’en de nombreux cas les VTR utilisées ne sont pas appropriées.
Pesticides organochlorés
Les pesticides organochlorés (POCs) sont des insecticides et pour la plupart des polluants environnementaux persistants (polluants organiques persistants ou POPs). Le DDT fut le premier POC utilisé à grande échelle aux États-Unis. Ces molécules qui persistent dans l’environnement, suscitent de vives inquiétudes de la part des pouvoirs publics car elles sont généralement lipophiles et bioaccumulables le long de la chaîne trophique. Malgré l’interdiction d’application de la plupart des POCs depuis longtemps dans un grand nombre de pays, leurs résidus ou leurs produits de dégradation pourraient avoir des impacts sur l’Homme (y compris la survenue de certains cancers) et son écosystème (Miligi et coll., 2006
; Chen et coll., 2007
).
Dieldrine
La dieldrine, stéréoisomère de l’endrine, appartient à la famille chimique des hydrocarbures chlorés non systémiques (ne pénétrant pas dans les tissus de la plante et non véhiculés par la sève) qui a été utilisé comme alternative au DDT. Elle agit par contact et ingestion (e-Pesticide Manual, 2004). En France, l’emploi de la dieldrine est interdit en agriculture depuis les années 1970 (arrêté du 2 octobre 1972). Encore employée comme insecticide non agricole, notamment dans la lutte contre les insectes xylophages et les termites, sa mise sur le marché est interdite et son utilisation règlementée par décret du 2 octobre 1992. La dieldrine est aujourd’hui interdite dans la plupart des pays, elle est l’une des douze premières molécules bannies par la Convention de Stockholm sur les polluants organiques persistants (POP).
Facilement absorbée par les voies cutanée, digestive et pulmonaire, la dieldrine s’accumule dans les tissus graisseux de l’organisme, à partir desquels peut s’effectuer une redistribution lente et progressive (Fiche Ineris, 2004).
Les premiers signes de toxicité aiguë concernent l’hyperexcitabilité neuromusculaire avec irritabilité accrue, tremblements et convulsions. En chronique, la toxicité la plus importante concerne le foie, toxicité caractérisée par une infiltration graisseuse du tissu et une prolifération du réticulum endoplasmique des cellules du parenchyme hépatique.
Chez l’Homme, l’intoxication aiguë se manifeste par la survenue de secousses musculaires puis d’un coma convulsif. De rares cas d’atteintes hépatiques ou rénales ont été signalés. À long terme, les personnes exposées à la dieldrine présentent un syndrome équivalent à une épilepsie idiopathique réversible, des atteintes du système nerveux périphérique, une fréquence plus élevée des atteintes hépatiques cliniques ou subcliniques (cirrhose portale) et une augmentation de la fréquence des irritations des bronches et des dermatoses de contact.
Pour le Circ, la dieldrine ne peut être classée du point de vue de sa cancérogénicité pour l’homme (groupe 3) (Circ, 1987
). Pour l’US-EPA
6
, la dieldrine est probablement cancérogène pour l’homme (classe B2). Il existe des preuves suffisantes chez l’animal et des preuves non adéquates chez l’homme. La dieldrine a été examinée mais n’est pas classée génotoxique par l’Union Européenne (catégorie 3, JOCE, 1993).
C’est un composé hépatocarcinogène non-génotoxique avéré chez la souris (Stevenson et coll., 1999
). Aucune étude n’a été menée sur le lien entre l’exposition par inhalation / contact cutané à la dieldrine et le développement de cancer. De nombreuses études par ingestion mettent en évidence une réponse de la souris à une exposition prolongée à la dieldrine différente de celle des autres espèces. En effet, l’hépatomégalie observée dans de nombreuses espèces est suivie par le développement de tumeurs (adénomes, carcinomes) hépatiques uniquement chez la souris (Davis et Fitzhugh, 1962
; Epstein, 1975
; NCI, 1978
; Reuber, 1980
). Des études réalisées sur des souris de différentes souches (Balb/c, CF1, B6C3F1, C3HeB/Fe, C3H/He et C57BL/6J) exposées à 0,65-1,3 mg de dieldrine/kg de p.c. par jour pendant 80-85 semaines mettent en évidence une augmentation de l’incidence de carcinomes et d’adénomes hépatocellulaires (Thorpe et Walker, 1973 ; NCI, 1978
; Tennekes et coll., 1982
; Meierhenry et coll., 1983
). La dieldrine agirait comme un promoteur de tumeur chez les souris mais pas chez les rats (Kolaja et coll., 1996
).
Les tests de mutations sur micro-organismes ont donné des résultats négatifs. Cependant, des lésions de l’ADN ont été mises en évidence sur cultures de cellules embryonnaires humaines
in vitro (US EPA, 2003a
) et sur cellules de la moelle osseuse de souris
in vivo (Majumdar et coll., 1976
). Le caractère mutagène de cette substance paraît donc probable. Les études menées
in vivo montrent que le traitement à la dieldrine entraîne une augmentation de la synthèse d’ADN et du taux de mitoses dans les hépatocytes de souris mais pas de rat (Stevenson, 1999
; Kamendulis et coll., 2001
). Le taux d’apoptose n’est pas modifié dans ces études. Des données contradictoires existent concernant les familles de CYPs induites (CYP1A, CYP1B, CYP2B) (Nim et Lubbet 1995
; Badawi et coll., 2000
). Il apparaît en outre, que comme la quasi totalité des pesticides organochlorés, la dieldrine est un composé activateur du récepteur nucléaire PXR (Coumoul et coll., 2002
).
Différentes études ont suspecté que le stress oxydant généré par la dieldrine est responsable de sa toxicité (Stevenson et coll., 1999
). En effet, le métabolisme de ce POC au niveau des hépatocytes génère des DROs, probablement par l’intermédiaire du cycle catalytique des cytochromes P450. Ce fait pourrait expliquer la variabilité inter-espèce observée au niveau du potentiel pro-carcinogène de cette molécule. En effet, chez la souris, les niveaux de systèmes anti-oxydants sont beaucoup plus faibles que chez le rat et seraient donc moins à même de protéger les hépatocytes des DROs générés.
En résumé, la dieldrine est classée 2B par l’US-EPA. Bien que considérée comme non génotoxique, la dieldrine produit des lésions de l’ADN dans des cellules embryonnaires humaines in vitro et dans des cellules de la moelle osseuse de souris in vivo. Elle semble agir comme promoteur de tumeur chez les souris mais pas chez les rats. Cette différence pourrait être attribuée aux systèmes anti-oxydants moins performants chez la souris qui protègent moins leurs hépatocytes des DRO, car la toxicité de la dieldrine pourrait être liée au stress oxydant qu’elle génère.
Lindane
Le lindane (γ-HCH) est l’un des isomères de l’hexachlorocyclohexane (HCH) synthétisé à partir du benzène et du chlore. Ce pesticide organochloré est utilisé pour de nombreuses applications agricoles, le traitement du bois ainsi qu’en médecine vétérinaire et humaine. Sa présence dans l’environnement est uniquement d’origine anthropique (utilisation et rejets indirects de fabrication ou de stockage). Il se retrouve aussi bien dans l’atmosphère que dans les eaux de surface ou souterraines. De part ses propriétés physico-chimiques, le lindane est persistant dans l’environnement, relativement stable dans l’atmosphère, se transporte sur de longues distances (régions d’émissions aux zones où il n’est ni produit, ni utilisé) et il est bioaccumulable le long de la chaîne trophique. Dans ce sens, il est considéré comme un Polluant Organique Persistant (POP) et a été ajouté en annexe de la convention de Stockholm lors de la conférence de Genève en 2009.
Chez l’Homme, le lindane est rapidement absorbé au niveau du tractus gastro-intestinal et distribué dans l’ensemble des tissus de l’organisme (tissus graisseux, cerveau, reins, muscles, poumons, cœur, rate foie et la circulation sanguine) et peut traverser la barrière placentaire (Baumann et coll., 1980
; Siddiqui et coll., 1981
). Métabolisé essentiellement au niveau hépatique selon quatre réactions enzymatiques (déshydrogénation, déshydrochloration, déchloration et hydroxylation), il génère des dérivés di-, tri-, tetra-, penta-, et hexachlorés.
Chez la souris et le rat, les premiers signes cliniques rapportés sont (pour une exposition par voie orale), une hypoactivité, une ataxie, une dyspnée et des convulsions. De plus, une étude menée chez la souris a montré que le lindane entraîne une diminution dose-dépendante des cellules de la moelle osseuse, des granulocytes progéniteurs de macrophages et des cellules souches pluripotentes de la moelle osseuse (Hong et Boorman, 1993
).
Chez l’homme, des doses létales de 180 à 300 mg/kg ont été rapportées. Des troubles neurologiques ont été décrits chez des enfants exposés à seulement 50 mg de lindane. L’effet le plus souvent rapporté en cas d’intoxication au lindane (ingestion accidentelle ou intentionnelle des insecticides, lotions ou nourriture) est une atteinte neurologique dont le principal signe clinique est la convulsion (Storen, 1955
; Harris et coll., 1969
; Starr et Clifford, 1972
; Munk et Nantel, 1977
; Davies et coll., 1983
). On observe également des nausées et des vomissements, une coagulation intra-vasculaire disséminée, des faiblesses musculaires et une nécrose des membres (Munk et Nantel, 1977
; Sunder Ram Rao et coll., 1988
). Au niveau de sa toxicité chronique, le lindane exerce des effets avérés neurotoxiques et hématologiques chez l’Homme.
In vivo, les animaux de laboratoire présentent une neurotoxicité, une immunotoxicité, une atteinte rénale et des atteintes hépatiques.
Les HCH techniques (en mélange α, β et γ) sont classés par le Circ en groupe 2B. L’EPA considère le lindane comme cancérogène possible chez l’Homme (groupe B2/C). Les effets cancérogènes de cette molécule ont été évalués au cours de 6 études réalisées chez la souris mais les données sont contradictoires. L’exposition par voie orale induit la survenue de tumeurs hépatiques bénignes et malignes chez les animaux des deux sexes au cours de deux études. Les résultats des autres études mentionnent un phénomène d’hépatotoxicité. Chez le rat, deux études décrivent également des processus d’hépato-cancérogénicité (Circ, 1979
).
Il ressort de toutes les études menées
in vivo, que le foie est l’organe le plus sensible au lindane, phénomène caractérisé par une hypertrophie, une stéatose et la mort des cellules par nécrose, phénomène essentiellement localisé au niveau des zones périportales (Junqueira et coll., 1988
; Videla et coll., 1990
). Plusieurs travaux ont imputé son effet hépatotoxique à sa capacité d’induire les CYPs et à son pouvoir pro-oxydant (Videla et coll., 2000
; Fernandez et coll., 2003
). Chez le rat, le lindane accroît les concentrations des CYPs, phénomène accompagné d’une production accrue de l’anion superoxyde dans les microsomes hépatiques, d’une induction de l’activité superoxyde dismutase cytoplasmique et de la peroxydation lipidique.
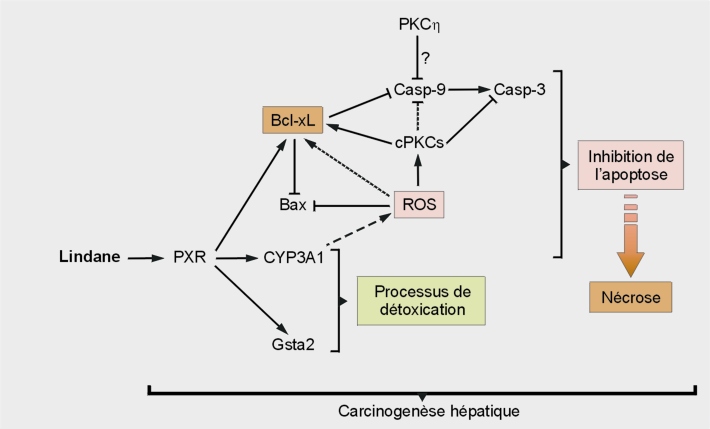
Sur un plan mécanistique, ce pesticide organochloré active le PXR, entraînant la surexpression du CYP3A4 chez l’Homme (CYP3A1 chez le rat) et des protéines anti-apoptotiques Bcl-2 et Bcl-xL (Lemaire et coll., 2004a
; Zucchini et coll., 2005
; Zucchini-Pascal et coll., 2009
). L’inhibition de l’apoptose qui en résulte s’accompagne d’une perturbation du mécanisme d’autophagie (voie majeure du catabolisme cellulaire permettant de dégrader et recycler les éléments endogènes détériorés). Ces deux phénomènes sont responsables de la mort des hépatocytes par nécrose (Zucchini-Pascal et coll., 2009
). Cette mort brutale entraînant une inflammation et un renouvellement cellulaire, constitue un facteur de prédisposition au développement des hépatocarcinomes. De plus, il semblerait que tous ces processus soient dépendants du pouvoir pro-oxydant du lindane. En effet, des données récentes démontrent que l’inhibition des processus apoptotiques, en partie responsable des phénomènes de nécrose au niveau des hépatocytes de rat est imputable à l’activation des protéines kinase C (PKCs). Ces médiateurs cellulaires seraient activés par les dérivés réactifs de l’oxygène (DRO) produits lors des processus de métabolisation dépendant des CYPs (modèle en figure 21.3
) (Zucchini-Pascal et coll., 2011
).
En résumé, le lindane est classé comme cancérogène possible (Circ et US-EPA). Le foie est considéré comme l’organe le plus sensible. Les propriétés pro-oxydantes du lindane, par induction de cytochromes P450, pourraient être impliquées dans l’inhibition des processus apoptotiques, favorisant par opposition la nécrose.
Endosulfan
Commercialisé à partir des années 1950, l’endosulfan (deux isomères alpha et bêta) est utilisé en agriculture pour le contrôle de divers insectes sur des cultures allant du café à la pomme de terre. Cette molécule comporte plusieurs atomes de chlore avec un complément soufré qui le différencie des autres produits de la famille des organochlorés. Non compatible avec les produits alcalins, il est parfois associé à d’autres pesticides : diméthoate, malathion, parathion, triazophos, monocrotophos, pirimicarb, augmentant son pouvoir toxique.
Ce POC, prohibé en 2005 par l’Union Européenne et en avril 2011 par la convention de Stockholm, sera effectivement totalement interdit à toute utilisation en 2012, avec des dérogations concernant certaines utilisations pendant 5 ans. Au même titre que la plupart des molécules de cette famille chimique, l’endosulfan est très controversé en raison de sa toxicité aiguë, son potentiel bioaccumulable, son pouvoir de perturbateur endocrinien et sa persistance dans l’environnement.
L’endosulfan est l’un des pesticides les plus toxiques actuellement sur le marché, responsable d’un grand nombre de décès par empoisonnement. Il est reconnu comme neurotoxique, quelle que soit l’espèce. L’EPA aux États-Unis le classe en catégorie I (hautement toxique), alors que l’organisation mondiale de la santé l’a placé dans la classe II (modérément dangereux). Les premiers signes cliniques d’intoxication sont : hyperactivité, tremblements, convulsions, perte de la coordination des mouvements, difficultés respiratoires, nausées et vomissements. Des doses de 35 mg/kg sont létales chez l’Homme et des doses sub-létales peuvent entraîner des dommages cérébraux irréversibles. De nombreuses études menées
in vitro et
in vivo le classent comme perturbateur endocrinien de par ses effets toxiques au niveau du développement et de la reproduction (notamment chez les mâles) et ses activités anti-androgéniques (Wilson et coll., 1997
; Andersen et coll., 2002
; Lemaire et coll., 2004b
; Orton et coll., 2011
).
Les effets de cet organochloré sur les processus de tumorigenèse sont très débattus et il n’est d’ailleurs pas classé par le Circ, l’EPA ou d’autres agences. Toutefois, des études menées
in vitro suggèrent des mécanismes d’action impliqués dans le développement et la progression tumorale. En effet, des doses sub-létales d’endosulfan (ainsi que ses métabolites) induisent des dommages à l’ADN et des mutations (Bajpayee et coll., 2006
; Antherieu et coll., 2007
; Silva et Beauvais, 2010
). De plus, le caractère génotoxique de ce pesticide a été démontré sur les cellules de la lignée HepG2 (cellules issues d’hépatocarcinomes) classiques ou exprimant les différents CYPs (Hashizume et coll., 2010
; Li et coll., 2011
).
Des données de 1978 issues du NCI (
National Cancer Institute) concernant l’endosulfan ont fait l’objet d’un réexamen. Ainsi, les études menées chez les rats décrivaient des lymphosarcomes chez les mâles et les femelles et des tumeurs du système reproducteur chez les femelles (Reuber, 1981
). Des tumeurs hépatiques ont également été observées chez les souris femelles traitées à l’endosulfan (Reuber, 1981
).
Au travers des différentes études publiées, les mécanismes de toxicité seraient similaires à ceux du lindane. L’endosulfan est générateur de stress oxydant qui serait à l’origine de la cytotoxicité observée au niveau hépatique (El-Shenawy, 2010
; Mor et Ozmen, 2010
). De plus, comme la majorité des organochlorés, il active le récepteur nucléaire PXR, entraînant une induction des CYP3A4 et 2B6 chez l’homme (Lemaire et coll., 2004b
; Casabar et coll., 2010
). En outre, son effet sur les processus apoptotiques semblent dépendre du tissu et du type cellulaire. Ainsi, ce composé inhibe l’apoptose induite et spontanée dans les cellules épidermiques HaCat et l’induit dans les cellules de la lignée HepG2. Des doses sub-létales de ce pesticide entraînent une hépatotoxicité corrélée à une nécrose et un état inflammatoire consécutifs au stress oxydant généré (Omurtag et coll., 2008
). Ce statut pourrait constituer un état de prédisposition au développement d’hépatocarcinomes.
En résumé, l’endosulfan qui n’est pas classé à ce jour, est suspecté de présenter des propriétés génotoxiques en particulier sur les cellules de la lignée HepG2. La plupart des études évoquent comme pour le lindane, un stress oxydant à l’origine de la cytotoxicité hépatique.
Chlordane
Ce pesticide organochloré non systémique fut utilisé intensivement entre 1950 et 1970, pour traiter les cultures, les pelouses, les jardins, les forêts et lutter contre les termites. De par sa persistance, sa bioaccumulation et sa forte toxicité pour les organismes vivants, il fut inclus dans la première liste des POPs à la convention de Stockholm. Seule la Chine était encore autorisée à en produire pour usage termiticide dans les habitations.
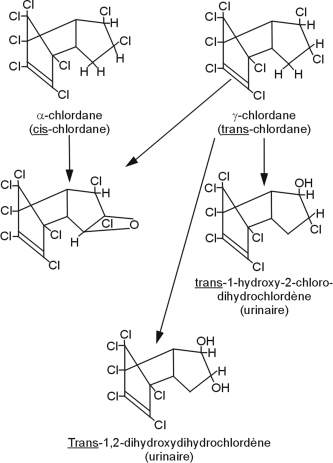
Le chlordane est absorbé‚ quelle que soit la voie d’exposition (inhalation, ingestion, contact cutané). Sa métabolisation est très lente avec la génération de métabolites pour la plupart moins toxiques que le produit parent à l’exception de l’oxychlordane, plus toxique que le chlordane et qui peut se stocker dans les graisses. Ce POC est métabolisé par deux voies de biotransformation, générant des époxydes et des radicaux libres responsables des phénomènes d’hépatotoxicité liés à ce composé (Cassidy et coll., 1994
) (figure 21.4
).
Chez l’Homme, l’exposition accidentelle à de fortes doses de chlordane par inhalation entraîne des douleurs thoraciques, des difficultés respiratoires, de la tachycardie, une perte de la coordination, l’engourdissement et/ou des effets gastro-intestinaux (douleurs, diarrhée, nausées et vomissements) (US EPA, 1980
). Quatre cas de décès ont été répertoriés et imputés à une intoxication au chlordane (Ineris, DRC-05-DR187, 2008). Ce composé est neurotoxique, immunotoxique, hépatotoxique et probablement cancérogène pour l’homme (groupe 2B, Circ).
Des études
in vivo ont permis de caractériser le chlordane comme agent promoteur tumoral au niveau hépatique. Chez la souris et le rat, ce composé augmente significativement l’incidence de nodules hyperplasiques hépatiques et de carcinomes hépatocellulaires. Il induit également des tumeurs au niveau de la thyroïde (néoplasmes folliculaires) et de la peau (hystiocytomes) (Epstein, 1976
; US NAS, 1977
; Telang et coll., 1982
), mais ne semble pas posséder de pouvoir génotoxique.
Le chlordane est un activateur du PXR et du CAR (Coumoul et coll., 2002
; Lemaire et coll., 2006
). Une étude a démontré que les phénomènes d’induction des CYPs, d’hépatomégalie, de prolifération des hépatocytes induits par le chlordane étaient absents chez des souris KO pour le PXR et le CAR (Ross et coll., 2010
). De plus, les lésions hépatocellulaires provoquées par le chlordane présentent des taux en Bcl-2 et Bcl-xL élevés, confirmant un mécanisme d’inhibition des processus apoptotiques (Christensen et coll., 1999
).
En résumé, le chlordane, classé 2B par le Circ est considéré d’après les études in vivo comme agent promoteur tumoral au niveau hépatique. Le développement des lésions pré-cancéreuses et cancéreuses par le chlordane pourrait résulter d’une activation de récepteurs nucléaires (PXR et CAR), phénomène responsable, au moins en partie de l’inhibition de l’apoptose et d’un état de prolifération cellulaire au niveau hépatique.
Chlordécone
Le chlordécone, insecticide organochloré, est un polluant organique persistant. L’exposition à cet organochloré peut s’effectuer par voie alimentaire, inhalation ou épidermique. Sa structure globale contenant 10 atomes de chlore lui confère une forte hydrophobicité et bioaccumulation en particulier au niveau hépatique et du tissu adipeux. Celle-ci est toutefois nettement inférieure au regard des autres dérivés organochlorés, de même que sa demi-vie qui diffèrent selon les espèces (120-160 j chez l’homme) versus plusieurs années pour les autres OC. Sa fonction cétone est à l’origine, chez quelques espèces incluant l’Homme, de sa métabolisation par hydratation suivie de sa glucuronoconjugaison puis de son élimination par voie biliaire, incluant un cycle entérohépatique. Une variabilité inter-espèce de ces processus, notamment de la formation du dérivé alcool, est répertoriée, en particulier entre les rongeurs et l’Homme.
Compte tenu des rapports récents sur cette molécule (Multigner et coll., 2007
; US EPA, 2009
; INVS, 2009
), seul un résumé des études toxicologiques ayant abouti à l’établissement des VTR et à l’évaluation des risques sanitaires sera présenté. L’essentiel des données de cancérogenèse provient de trois sources majeures.
D’une part, des observations et examens cliniques approfondis effectués après la survenue d’une exposition, principalement professionnelle dans une fabrique de chlordécone aux États-Unis à Hopewell (Cannon et coll., 1978
; Cohn et coll., 1978
; Guzelian, 1992
). Lorsque l’exposition s’est traduite par une charge corporelle dépassant 1 mg/l de chlordécone dans le sang, elle a entraîné un ensemble de symptômes et de signes cliniques regroupés sous la dénomination du « syndrome du Kepone » (nom commercial du chlordécone). Ce syndrome se caractérise, principalement par des troubles neurologiques (tremblements des membres, incoordination motrice, troubles de l’humeur et de la mémoire récente…) accompagné d’une hépatomégalie sans lésions cancéreuses. La sévérité du tableau clinique corrélait à la concentration circulante en chlordécone. Ces signes et symptômes sont progressivement réversibles lorsque l’exposition cesse. Après un suivi de plus de 10 ans, aucune augmentation d’une quelconque pathologie tumorale n’a été identifiée dans cette population de travailleurs exposés. Par ailleurs, une étude épidémiologique menée auprès des populations antillaises a montré que la concentration plasmatique en chlordécone est associée de manière dose-dépendante à un risque augmenté de survenue du cancer de la prostate. Selon les auteurs, la plausibilité biologique de ces associations résiderait dans les propriétés œstrogéniques de la molécule (Multigner et coll., 2010
).
D’autre part, des études expérimentales ont été réalisées chez les rongeurs principalement (NCI, 1976
; Larson et coll., 1979
; Sirica et coll., 1989
). La première (NCI, 1976
) conclut à une hyperplasie hépatique et une augmentation des hépatocarcinomes, avec une fréquence plus élevée chez les rats femelles que les mâles et l’inverse chez la souris. Ces tumeurs, non retrouvées dans 15 autres organes analysés, sont bien circonscrites, différenciées et peu vascularisées. Quoiqu’avec des effectifs plus faibles et des résultats moins tranchés, l’étude de Larson et coll. (1979
) menée chez le rat confirme la précédente. Cette dernière attribue au chlordécone un rôle de promoteur de cancérogenèse hépatique. Par ailleurs, quelle que soit l’espèce, le chlordécone entraîne les signes du syndrome du Kepone (syndrome clinique caractérisé principalement par des troubles neurologiques accompagnés d’une hépatomégalie et d’une altération de certaines caractéristiques spermatiques chez les mâles). Cependant, la conclusion de ces travaux en termes de risques sanitaires est délicate du fait, soit que les doses d’exposition dans certaines de ces études expérimentales excèdent fortement celles des travailleurs d’Hopewell (chez qui aucune atteinte tumorale hépatique n’a été constatée après un suivi de plus de 10 ans), soit que les témoins historiques présentent une fréquence de tumeurs anormalement élevée par rapport à la norme admise chez la souris. Des résultats publiés par le
National Toxicology Program Institute of Health (NTP, 1990
) sur un analogue du chlordécone, le mirex, confirme la non-génotoxicité et l’hépatotoxicité de ce dernier, mais contredit en partie certains de ses effets cancérogènes
in vivo (absence d’effet lié au sexe, foyers tumoraux dans d’autres tissus et absence d’hépatocarcinome).
Enfin, des investigations conduites au niveau cellulaire montrent le caractère non génotoxique (Galloway et coll., 1987
; Mortelmans et coll., 1998
) et œstrogéno-mimétique de cette molécule, avec la particularité d’avoir des propriétés agonistes vis-à-vis de l’ERα, et antagonistes vis-à-vis de l’ERβ (Palmiter et Mulvihill, 1978
; Hammond et coll., 1979
; Kocarek et coll., 1994
; Lemaire et coll., 2006
). Ces données pourraient rendre compte de son potentiel promoteur tumoral décrit
in vivo. Par ailleurs, à l’instar du phénobarbital, promoteur de cancérogenèse connu provoquant une hyperplasie et une hépatomégalie, le chlordécone provoque l’induction des cytochromes P450, CYP3A4 et 2B6 hépatocytaires tout en transactivant le récepteur nucléaire PXR (Lemaire et coll., 2004a
). Enfin, cette molécule a été décrite comme inhibant les jonctions cellulaires et réprimant certaines protéines impliquées dans l’adhésion cellulaire, telle que la β caténine, dans des cellules épithéliales mammaires (Starcevic et coll., 2001
), caractéristiques de composés favorisant la progression et d’invasion tumorale. Cet effet n’a cependant pas été signalé lors des études
in vivo.
S’agissant de l’évaluation des risques sanitaires, le chlordécone étant dénué de génotoxicité, est considéré par les agences comme présentant un seuil d’action dose-dépendant (voir paragraphe « Estimation des risques »). Les effets critiques retenus pour l’élaboration des Valeurs de Références Toxicologiques ne concernent pas les effets cancérogènes, car ceux-ci sont retrouvés chez les rongeurs seulement, pour des doses élevées, donc non extrapolables à l’Homme. Plus largement, les études laissent apparaître que les effets trouvés chez l’Homme (hépatomégalie, induction enzymatique notamment) sont reproduits chez les rongeurs, mais non l’inverse (forte hépatotoxicité, follicules néoplasiques rongeurs-spécifiques). Le Circ a ainsi classé ce composé dans le groupe 2B (cancérogène possible) (Circ, 1987
). Une VTR par exposition orale chronique de 0,0005 mg.kg
-1.j
-1 (NOAEL/100) a donc été estimée en prenant les altérations rénales, comme effets critiques. Soulignons, qu’en dépit des fortes expositions des travailleurs de Hopewell, aucun symptôme ou signe de la sphère rénale n’a été observé. La VTR relative à l’exposition aiguë considérant des effets critiques neuronaux est de 0,01 mg.kg
-1.j
-1 (Afssa, 2007
).
En résumé, le chlordécone classé 2B par le Circ et non génotoxique est surtout associé au cancer de la prostate d’après l’étude épidémiologique menée auprès des populations antillaises. Les propriétés œstrogéniques de la molécule pourraient être impliquées dans cette association.
Pesticides organophosphorés
La plupart des données toxicologiques relatives à ces molécules ont été rapportées dans une monographie du Circ (Circ, 1983
). Les travaux complémentaires et postérieurs à cette monographie sont analysés.
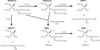
Malathion
Cet insecticide organophosphoré (OP) à action non systémique agit par ingestion, contact ou inhalation. Son principal mode d’action est l’inhibition de l’acétylcholinestérase. Au plan pharmacocinétique, le malathion est efficacement absorbé par le tractus gastro-intestinal des mammifères, biotransformé puis rapidement excrété par voie urinaire. Ses effets toxiques sont attribués à sa désulfuration oxydative conduisant à la formation du malaoxon au niveau du foie. À faibles doses d’exposition, la formation du malaoxon impliquerait le CYP1A2 tandis qu’à doses plus élevées sont impliqués les CYP2B6 et 3A4 (Buratti et coll., 2005
). Le malathion et le malaoxon sont également hydrolysés et détoxifiés par les carboxylestérases, essentiellement au niveau du foie. Des réactions de déalkylation, probablement sous la dépendance des glutathion-S-transférases contribuent vraisemblablement aussi à cette élimination (figure 21.5
).
L’hydrolyse enzymatique significativement plus lente du malathion et du malaoxon par les carboxylestérases d’insectes, rend compte de leurs spécificités d’action par rapport aux mammifères. La toxicité du malathion est de ce fait potentialisée par tout composé capable d’inactiver les carboxyestérases (impuretés techniques, autres esters OP). Le malaoxon constitue à la fois un inhibiteur et un substrat des carboxylestérases mais sa détoxication par hydrolyse apparaît la réaction prédominante.
Les tests réglementaires sur bactéries, levures et drosophiles ne montrent aucun potentiel mutagène du malathion. Il en est de même pour les tests de mutation dominante létale chez la souris et le test UDS (
Unscheduled DNA Synthesis), qui mesure les capacités de réparation de l’ADN
in vivo en réponse aux toxiques. Le malathion provoque cependant un accroissement de fréquence d’échanges de chromatides sœurs sur cellules de mammifères ainsi que d’aberrations chromosomiques tant
in vitro qu’
in vivo chez la souris. Les études expérimentales sur le malathion et le malaoxon menées chez l’animal ne montrent aucun potentiel cancérogène, ni tératogène ou embryotoxique. Les données épidémiologiques et toxicologiques chez les populations exposées faisant défaut lors de son évaluation par le Circ en 1987
, cette molécule est considérée de ce fait comme étant non génotoxique et non-cancérogène (groupe 3 du Circ). Certaines études récentes conduites sur la lignée d’hépatome humain HepG2 d’origine humaine, montrent cependant que le malathion provoque une cytotoxicité, un stress oxydant significatif (mesure de la peroxydation lipidique) et une génotoxicité (essai des Comètes). Son potentiel hépatotoxique a également été démontré chez le rat (Kalender et coll., 2010
; Moore et coll., 2010
).
En résumé, le malathion, considéré pourtant comme non génotoxique et non cancérogène par le Circ en 1987
, présente une certaine génotoxicité
in vitro sur cellules de mammifères,
in vivo chez la souris et plus récemment sur la lignée d’hépatome humain HepG2 ainsi qu’un potentiel hépatotoxique.
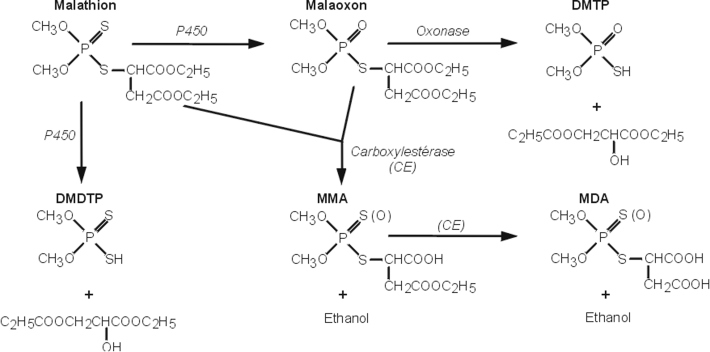
Parathion
Utilisé pour traiter les sols et les parties aériennes des végétaux, cet organophosphoré (OP) cible également l’acétylcholinestérase. Il est rapidement absorbé par voie épidermique, respiratoire, digestive et oculaire, puis éliminé essentiellement par voie rénale. Le parathion est principalement biotransformé au niveau du foie mais aussi dans d’autres tissus (cerveau, poumons…). Ses effets toxiques sont liés à sa désulfuration oxydative par les cytochromes P450, CYP2B6 et 2C19 aboutissant à la production du paraoxon (Foxenberga et coll., 2011
). Ces mêmes enzymes conduisent à sa détoxication via la formation d’alkylphosphates et de p-nitrophénol, qui excrétés par voie urinaire constituent des biomarqueurs d’exposition chez les applicateurs. Le paraoxon est quant à lui détoxifié par les estérases et la paraoxonase 1 (PON1) (figure 21.6
).
Concernant la génotoxicité, les tests réglementaires se révèlent négatifs, que ce soit en présence ou en absence de systèmes d’activation métabolique. En termes de cancérogenèse, certaines études menées par voie orale chez les rats mâles et femelles montrent un accroissement significatif et dose-dépendant des tumeurs cortico-surrénaliennes, essentiellement des adénomes. Chez les mâles, des carcinomes du pancréas et des adénomes folliculaires de la thyroïde sont observés. Ces données ne sont pas confirmées par d’autres études menées chez le rat et les résultats obtenus chez la souris (voie orale) ne montrent aucune augmentation significative du nombre de tumeurs. La difficulté d’interprétation de ces études, du fait des courtes périodes d’exposition, du faible nombre d’animaux et de tissus examinés, ainsi que du nombre réduit de tumeurs, a conduit le Circ à classer le parathion dans le groupe 3 (« Inclassable quant à sa cancérogénicité pour l’homme »). Au total, les études épidémiologiques chez les populations exposées faisant défaut, cette molécule est de ce fait considérée comme non génotoxique et non cancérogène. Cependant, à l’instar du malathion, des études récentes menées sur la lignée d’hépatome HepG2 démontre que le parathion (ainsi que le méthyl-parathion) induit un stress oxydant (mesure de la peroxydation lipidique) et une génotoxicité (essai des Comètes) significatifs (Unaldi Coral et coll., 2009
; Edwards et coll., 2011
).
En résumé, le parathion est considéré comme non génotoxique et non cancérogène par le Circ. Cependant, comme pour le malathion, des études récentes évoquent l’apparition d’un stress oxydant et des effets génotoxiques associés à son exposition.
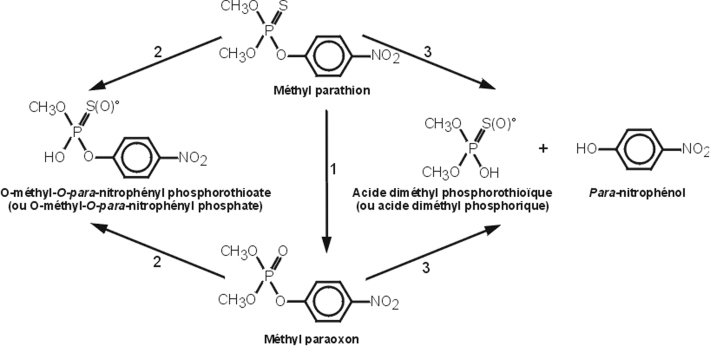
Méthyl-parathion
Cet insecticide de contact, agissant également par voie digestive, est comme les précédents composés organo-phosphorés, un inhibiteur de l’activité cholinestérase. Facilement absorbé par voie orale, épidermique et respiratoire, il se distribue de façon très rapide dans les tissus de l’organisme, puis est majoritairement éliminé par les urines. Ses principaux métabolites formés au niveau du foie sont le para-nitrophénol, le diméthyl phosphate et le méthylparaoxon formé quelques minutes après son administration. Ce dernier, inhibiteur actif de l’acétylcholinéstérase, est responsable de sa toxicité (figure 21.7
).
Les réactions 2 et 3 font intervenir les glutathion alkyl et aryl transférases, respectivement, la formation du para-nitrophénol pouvant également être catalysée par des hydrolases microsomiales ou non microsomiales. L’excrétion urinaire du para-nitrophénol est un bioindicateur d’exposition à cet organophosphoré.
Le méthyl-parathion apparaît faiblement ou non mutagène dans les systèmes bactériens (diverses souches de
Salmonella typhimurium, en présence de systèmes biotransformant ou pas) et
Drosophila melanogaster, mais il l’est chez la levure. Cependant, dans plusieurs modèles de cellules de mammifères (V79 hamster chinois, cellules lymphoïdes humaines B35M), il induit l’échange de chromatides sœurs ainsi que certaines mutations, mais ne provoque ni d’aberrations chromosomiques, ni de synthèse non programmée de l’ADN (test UDS). Ceci est également vrai
in vivo chez la souris où les tests de mutation dominante létale et d’anormalités chromosomiques se révèlent négatifs. Il y a au total suffisamment d’arguments montrant que le méthyl-parathion est génotoxique dans nombre de systèmes cellulaires, mais non chez les organismes vertébrés. En ce qui concerne son potentiel cancérogène, aucun effet n’a été rapporté après son administration par voie orale chez les rongeurs, mais son caractère tératogène a été montré chez la souris, mais non chez le rat. Faute de données épidémiologiques et toxicologiques chez l’homme lors de son évaluation par le Circ en 1987
, le méthyl-parathion est classé comme molécule non génotoxique et non cancérogène (groupe 3 du Circ, 1987
). Des publications postérieures à ces évaluations démontrent néanmoins, que comme le malathion et le parathion, cette molécule est génératrice de stress oxydant et qu’elle présente un caractère génotoxique et pro-néoplasique avéré (Unaldi Coral et coll., 2009
; Edward et coll., 2013
).
En résumé, le méthyl-parathion classé comme non génotoxique et non cancérogène présente cependant des propriétés mutagènes chez la levure, une certaine génotoxicité dans divers modèles cellulaires. Des travaux récents, lui attribuent un caractère génotoxique et pro-néoplasique.
Autres pesticides
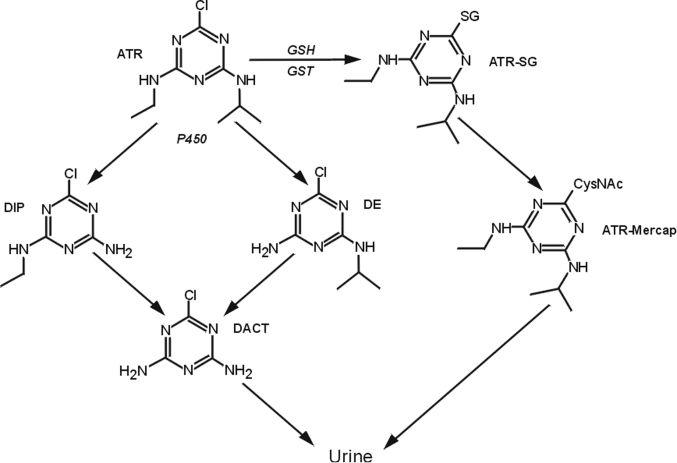
Atrazine
L’atrazine ou 2-chloro-4-(éthylamine)-6-(isopropylamine)-s-triazine, est un herbicide de la famille chimique des triazines (figure 21.8
). Interdite dans l’Union Européenne depuis 2004, l’atrazine est toujours utilisée dans un grand nombre de pays pour le traitement des mauvaises herbes dans de nombreuses cultures annuelles ou pérennes. C’est l’un des herbicides les plus couramment utilisés. L’atrazine inhibe le transport d’électrons nécessaire au mécanisme de la photosynthèse. Peu d’intoxications aiguës à l’atrazine ont été répertoriées chez l’Homme. Quelques cas cliniques d’inflammation cutanée ont été décrits. En outre, aucune manifestation toxique n’a été observée chez des personnes ayant ingéré intentionnellement (suicides) cet herbicide. Ces données semblent suggérer une apparente innocuité de l’atrazine à court terme (US EPA, 2003b
; Loosli, 1995
).
In vivo, ce composé entraîne chez les animaux de laboratoire des irritations modérées ou faible au niveau de la peau et des yeux, respectivement (US EPA, 2010
). Après ingestion, les signes d’intoxication concernent le système cardio-vasculaire (chien), le foie (rat, souris, cochon), les reins (rat et cochon) et le système endocrinien (rat). Il apparaît que la plupart des troubles observés après ingestion chronique d’atrazine concernent des altérations endocriniennes (action au niveau de l’hypophyse, métabolisme des stéroïdes). On note une dérégulation des niveaux d’hormone lutéïnisante, d’œstradiol, de progestérone et de prolactine chez le rat, créant chez les femelles une diminution de l’activité œstrogénique et donc une perturbation du cycle ovarien (infertilité, arrêt du développement embryonnaire). Cet herbicide entraîne également des troubles chez la descendance, avec des courbes de diminution de poids réduites chez les nouveau-nés dues à la réduction de la lactation (en particulier du taux de prolactine chez les mères) (Birnbaum et Fenton, 2003
).
Le lien entre atrazine et tumorigenèse chez l’homme fait encore débat à l’heure actuelle. Des études menées
in vivo chez les rats ont mis en évidence l’apparition précoce de tumeurs des glandes mammaires, de l’hypophyse, de l’utérus (Wetzel et coll., 1994
), et des atteintes hématopoïétiques (Mencoboni et coll., 1992
; Pinchuk et coll., 2007
). L’atrazine ne semble pas être génotoxique et son pouvoir de promoteur de tumeurs est imputé à ses capacités de perturbateur endocrinien (Devos et coll., 2003
).
En résumé, l’atrazine est considérée comme une molécule non génotoxique. Ses propriétés de perturbateur endocrinien pourraient être responsables des tumeurs précoces observées chez le rat.
Captane
Le captane appartient au groupe des phtalimides. Il est caractérisé par un anneau cyclohexane. Le captane est un dérivé fongicide non systémique à spectre large. Hormis son utilisation agronomique, cette molécule est utilisée pour ses propriétés dans les peintures, stabilisateurs, colles, laques, papiers, plastique, cuir, le savon, dans certains sprays vétérinaires… La grande majorité des tests classiques et réglementaires attestent que le captane présente une génotoxicité (test d’Ames, mutations sur plusieurs loci, aberrations chromosomiques, échanges de chromatides sœurs…). Les tests du micronoyau sur érythrocytes et d’anomalie chromosomique effectuée après traitement
in vivo chez les rongeurs, apparaissent négatifs et le test de mutations dominantes létales reste controversé. L’ensemble de ces données a été jugé insuffisant, notamment par l’EPA, pour classer ce composé comme génotoxique chez les mammifères. Le captane est donc considéré comme non-génotoxique présentant de ce fait un seuil d’action toxicologique dose dépendant
7
. Cet effet non-génotoxique n’est cependant pas prouvé à ce jour.
Par ailleurs, son administration répétée chez des souris conduit à une dérégulation de l’expression des cytochromes P450 hépatique et à une induction d’espèces pro-oxydantes. Les expériences complémentaires menées
in vitro démontrent que le mécanisme d’inhibition serait directement dû aux métabolites et non au composé parent (Paolini et coll., 1999
). Cela est confirmé par les données métaboliques (Berthet et coll., 2012
, voir figure 21.9
), conduisant l’EPA à supposer l’implication de métabolites hautement réactifs du captane, en particulier des dérivés thiophosgène et/ou tetrahydrophthalimide, dans son effet cancérogène. Ces métabolites sont formés et souvent retrouvés aux niveaux du duodénum, de l’urine et du sang, où ils pourraient exercer leurs effets délétères.
En termes de cancérogénicité, les études expérimentales conduites chez les rongeurs, indiquent, qu’administrée par voie alimentaire, cette molécule provoque des adénomes et carcinomes intestinaux (essentiellement au niveau de la partie proximale du duodénum) chez la souris et quel que soit le sexe, mais non chez le rat. Le captane est considéré par l’EPA comme cancérogène potentiel, mais uniquement à des doses élevées et chroniques, à l’origine d’une cytotoxicité et d’une hyperplasie cellulaire régénérative. Ces effets sont doses dépendantes et réversibles après exposition (US EPA, 2004
, 2005
). Compte tenu de ces mécanismes d’action, le captane serait non-cancérogène à de faibles niveaux de doses auxquelles les populations humaines seraient exposées. Sur ces bases, il n’a pu être classé par le Circ quant à sa cancérogénicité pour l’homme (groupe 3).
En résumé, le captane, considéré par l’EPA comme cancérogène potentiel à doses élevées et chroniques (non classé par le Circ), induit une cytotoxicité et une hyperplasie cellulaire régénérative.
En conclusion, l’objectif de cette analyse était de tenter de mettre en relation les mécanismes d’action cellulaires et moléculaires de molécules phytopharmaceutiques, avec certains de leurs effets cancérogènes avérés ou suspectés, notamment à partir des études épidémiologiques. Dans l’impossibilité d’être exhaustifs, la démarche proposée ici est fondée uniquement sur un certain nombre de molécules appartenant à diverses familles chimiques (organophosphorés, organochlorés…). Celles-ci ont été sélectionnées, d’une part en fonction des données épidémiologiques issues de l’Agricultural Health Study, d’autre part selon leurs tonnages annuels, enfin suivant leur classement en termes de cancérogénicité. Au niveau des mécanismes d’action, les publications de bonne qualité, traitant des processus cellulaires et moléculaires mettent en évidence des mécanismes d’altérations du matériel génétique, de déséquilibre des processus de survie et de mort cellulaires, de liaison à des récepteurs nucléaires et hormonaux vitaux, de bioactivation métabolique et de génération de stress oxydant. Ces informations ont pu être croisées avec les données toxicologiques réglementaires (essentiellement issues de l’expérimentation animale) et celles des agences d’évaluation des risques (indices de risque, valeurs toxicologiques de référence).
L’analyse bibliographique montre que la plupart des pesticides provoquent des perturbations cellulaires et moléculaires majeures : modification d’expression des cytochromes P450, glucuro- et glutathion-transférases, production d’espèces oxygénées (induction de NFkB, HSP70, c-fos, GADD153…), génotoxicité (pré- ou post-bioactivation), induction de cascades de MAP kinases…. Plusieurs (lindane, dieldrine, aldicarbe…) présentent des effets anti-apoptotiques avérés (inhibition des caspases, induction des protéines anti-apoptotiques de la famille Bcl-2…), liés aux cancers. Ces effets étroitement reliés, varient cependant entre les espèces, les individus et les tissus (foie, épiderme…) et sont souvent d’origine métabolique. Ces molécules interagissent de plus avec de nombreux récepteurs nucléaires/hormonaux humains gouvernant des voies de signalisation cellulaires physiologiquement primordiales. De nombreux composés organochlorés, pyréthrinoïdes, organophosphorés, activent en particulier le récepteur nucléaire PXR, régulant le métabolisme des hormones stéroïdes et des sels biliaires, tout en induisant les CYP3A4 et 2B6 (hépatocytes humains), placés sous son contrôle. Après fixation sur ce récepteur, le lindane dérégule en particulier l’expression de gènes impliqués dans le métabolisme des xénobiotiques (CYP, GST et GSTa2) et les voies de signalisation liées au stress (PKCs, eIF2b) ou à la mort cellulaire. Des données mécanistiques explicatives sont ainsi apportées quant aux effets hépatotoxiques et cancérogènes montrés in vivo chez le rat en réponse à cette molécule, et suspectés (mais non retrouvés) chez l’Homme.
De plus, le pouvoir antagoniste, notamment des organochlorés vis-à-vis du récepteur aux androgènes a été largement mis en évidence, la plupart activant également les récepteurs aux œstrogènes (ERa/b), avec des actions contraires selon les isoformes a et b. D’autres activent les récepteurs aux rétinoïdes RARb/g (mais non RARa et RXR), tout en induisant le CYP26A1 placé sous leur contrôle, résultat original d’importance toxicologique, puisque les RARs interviennent dans nombre de fonctions vitales (morphogenèse, métabolisme des rétinoïdes…). En ce qui concerne le récepteur d’aryl d’hydrocarbone AhR, il apparaît de façon inattendue, qu’à l’inverse des ligands prototypes (dioxine, hydrocarbures aromatiques polycycliques…), certains composés (2,4-D, carbaryl, thiabendazole…) sont capables d’induire les cytochromes P450 CYP1A1/2, sans emprunter cette voie de signalisation classique. D’autres molécules agissent également sur les cascades de MAP kinases, éléments clés des voies de transduction du signal intracellulaire, impliqués dans d’innombrables processus biologiques. Les travaux démontrent que plusieurs organochlorés (endosulfan, lindane…) activent les voies kinases (ERK1/2, JNK) et les PKCs, inhibent l’apoptose hépatocytaire (lindane) et s’avèrent génotoxiques sans bioactivation préalable (endosulfan). Le lindane perturbe en outre considérablement les processus d’autophagie tout en entraînant parallèlement une inhibition de l’apoptose hépatocytaire, attestée par la réduction durable de la protéine anti-apoptotique Bcl-xL, l’inhibition des activités caspases-3 et -9 et de la protéine pro-apoptotique Bax. La dérégulation de ces processus de protection cellulaire (apoptose et autophagie) aboutit in fine à une mort de type nécrotique, trois cascades signalétiques qui sont très étroitement inter-connectées. L’inhibition de l’apoptose et l’induction de la nécrose résultent de l’activation des PKCs, consécutive à leur pouvoir pro-oxydant. Plusieurs études toxicogénomiques confirment finalement que de nombreux pesticides provoquent des perturbations transcriptionnelles significatives de gènes impliqués dans d’innombrables fonctions biologiques. Celles-ci ainsi que les altérations phénotypiques et génotypiques précoces sont à notre sens très insuffisamment prises en considération dans les dossiers d’enregistrement.
Ainsi, l’apport des études mécanistiques cellulaires et moléculaires, qu’elles soient menées in vivo ou in vitro, apparaît essentiel dans l’estimation des risques sanitaires potentiels des xénobiotiques environnementaux. Ces mécanismes conditionnent la réaction au stress environnemental et déterminent la réponse adaptative de la cellule, et donc de l’organisme. La « décision » que prendra la cellule face à une agression chimique dépendra d’une quantité importante de paramètres allant de sa capacité à apprécier le danger (découlant de la variabilité inter-individuelle et/ou du polymorphisme génétique ou de réponse), des signaux mis en jeu, des facteurs de prédispositions présents dans l’organisme… La connaissance des paramètres génétiques, épigénétiques ou de signalisation impliqués dans le développement de tumeurs, mais également dans l’établissement d’un climat favorable au processus de cancérogenèse, apparaît donc primordiale dans l’évaluation des risques sanitaires. Dans ce cadre, la connaissance des processus moléculaires engagés est essentielle et permet de mettre en évidence de nouveaux biomarqueurs prédictifs témoignant d’une relation moléculaire entre l’exposition aux pesticides et les facteurs de cancérogenèse.
Pourtant, bien qu’indispensable, l’identification de tels mécanismes d’actions et de biomarqueurs in vitro ne suffit pas en elle-même, à prévoir la survenue de pathologies pesticides-dépendantes. Il est nécessaire de les confronter aux propriétés de réactivité chimique, de métabolisme, aux données toxicologiques animales, épidémiologiques… Le développement d’outils de gestion/traitement de données et de modélisation mathématique est primordial au succès de cette approche, où les modèles d’exposition, de relations dose-réponse, toxicocinétiques, de biologie systémique intégrant le polymorphisme de réponse aux toxiques tiennent une place prépondérante. Seule une telle approche intégrative de la réponse physiologique, peut in fine conduire à l’objectif d’une toxicologie prédictive, c’est-à-dire capable d’anticiper les propriétés toxicologiques des pesticides à partir d’expériences in vitro et in silico, ainsi que des données in vivo et épidémiologiques disponibles, en réduisant le recours systématique à l’animal, scientifiquement et éthiquement critiquable.
L’analyse des mécanismes d’action confirme cependant que les risques sanitaires liés à l’exposition à ces molécules doivent être reconsidérés avec attention. Elle souligne qu’en dépit des études toxicologiques réglementaires, aboutissant à la mise sur le marché des pesticides, un certain nombre de leurs effets biologiques chez l’Homme ne sont pas suffisamment pris en considération par la réglementation, surtout lors d’expositions à faibles doses, de longue durée et sous forme de mélanges. Le regroupement des données épidémiologiques, des données de toxicologie réglementaire et fondamentale permet néanmoins d’apporter un grand nombre d’arguments sur la plausibilité d’une relation entre pesticides et cancer.
Bibliographie
[1]AFSSA.Avis du 6 septembre 2007. Avis de l’Agence française de sécurité sanitaire des aliments relatif à l’actualisation des données scientifiques sur la toxicité du chlordécone en vue d’une éventuelle révision des limites tolérables d’exposition proposées par l’Afssa en 2003.
[http://www.afssa.fr/Documents/RCCP2007sa0305.pdf].

[2] ANDERSEN HR, VINGGAARD AM, RASMUSSEN TH, GJERMANDSEN IM, BONEFELD-JURGENSEN EC. Effects of currently used pesticides in assays for estrogenicity, androgenicity, and aromatase activity in vitro.
Toxicol Appl Pharmacol. 2002;
179:1
-12
[3]anonyme, Asbestos, asbestosis, and cancer: the Helsinki criteria for diagnosis and attribution.
Scand J Work Environ Health. 1997;
23:311
-316
[4] ANTHERIEU S, LEDIRAC N, LUZY AP, LENORMAND P, CARON JC, RAHMANI R. Endosulfan decreases cell growth and apoptosis in human HaCaT keratinocytes: partial ROS-dependent ERK1/2 mechanism.
J Cell Physiol. 2007;
213:177
-186
[5] BADAWI AF, CAVALIERI EL, ROGAN EG. Effect of chlorinated hydrocarbons on expression of cytochrome P450 1A1, 1A2 and 1B1 and 2- and 4-hydroxylation of 17beta-estradiol in female Sprague-Dawley rats.
Carcinogenesis. 2000;
21:1593
-1599
[6] BAJPAYEE M, PANDEY AK, ZAIDI S, MUSARRAT J, PARMAR D, et coll. DNA damage and mutagenicity induced by endosulfan and its metabolites.
Environ Mol Mutagen. 2006;
47:682
-692
[7] BAUMANN K, ANGERER J, HEINRICH R, LEHNERT G. Occupational exposure to hexachlorocyclohexane. I. Body burden of HCH-isomers.
Int Arch Occup Environ Health. 1980;
47:119
-127
[8] BERTHET A, BOUCHARD M, VERNEZ D. Toxicokinetics of captan and folpet biomarkers in dermally exposed volunteers.
J Appl Toxicol. 2012 [Epub 2011 Mar 5];
2(3):202
-209
[9] BIRNBAUM LS, FENTON SE. Cancer and developmental exposure to endocrine disruptors.
Environ Health Perspect. 2003;
111:389
-394
[10] BOYER B, VALLÈS AM, EDME N. Induction and regulation of epithelial-mesenchymal transitions.
Biochem Pharmacol. 2000;
60:1091
-1099
[11] BURATTI FM, D’ANIELLO A, VOLPE MT, MENEGUZ A, TESTAI E. Malathion bioactivation in the human liver: the contribution of different cytochrome p450 isoforms.
Drug Metab Dispos. 2005;
33:295
-302
[12] BUTTERWORTH BE, ELDRIDGE SR, SPRANKLE CS, WORKING PK, BENTLEY KS, HURTT ME. Tissue-specific genotoxic effects of acrylamide and acrylonitrile.
Environ Mol Mutagen. 1992;
20:148
-155
[13] CANNON SB, VEAZEY JMJR, JACKSON RS, BURSE VW, HAYES C, et coll. Epidemic kepone poisoning in chemical workers.
Am J Epidemiol. 1978;
107(6):529
-537
[14] CASABAR RC, DAS PC, DEKREY GK, GARDINER CS, CAO Y, et coll. Endosulfan induces CYP2B6 and CYP3A4 by activating the pregnane X receptor.
Toxicol Appl Pharmacol. 2010;
245:335
-343
[15] CASSIDY RA, VORHEES CV, MINNEMA DJ, HASTINGS L. The effects of chlordane exposure during pre- and postnatal periods at environmentally relevant levels on sex steroid-mediated behaviors and functions in the rat.
Toxicol Appl Pharmacol. 1994;
126:326
-337
[16] CHEN L, DING L, JIN H, SONG D, ZHANG H, et coll. The determination of organochlorine pesticides based on dynamic microwave-assisted extraction coupled with on-line solid-phase extraction of high-performance liquid chromatography.
Anal Chim Acta. 2007;
589:239
-246
[17] CHRISTENSEN JG, ROMACH EH, HEALY LN, GONZALES AJ, ANDERSON SP, et coll. Altered bcl-2 family expression during non-genotoxic hepatocarcinogenesis in mice.
Carcinogenesis. 1999;
20:1583
-1590
[18] CHRISTOFORI G. New signals from the invasive front.
Nature. 2006;
441:444
-450
[20]CIRC. Miscellaneous Pesticides. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Volume 30.
1983.
424 p.p.
[21]CIRC. Overall Evaluations of Carcinogenicity : An Updating of IARC Monographs Volumes 1 to 42. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (suppl 7).
1987.
440 p.p.
[22] COHN WJ, BOYLAN JJ, BLANKE RV, FARISS MW, HOWELL JR, GUZELIAN PS. Treatment of chlordecone (Kepone) toxicity with cholestyramine. Results of a controlled clinical trial.
N Engl J Med. 1978;
298:243
-248
[23] COUMOUL X, DIRY M, BAROUKI R, . PXR-dependent induction of human CYP3A4 gene expression by organochlorine pesticides.
Biochem Pharmacol. 2002;
64:1513
-1519
[24] DAVIS KJ, FITZHUGH OG. Tumorigenic potential of aldrin and dieldrin for mice.
Toxicol Appl Pharmacol. 1962;
4:187
-189
[25] DAVIES JE, DEDHIA HV, MORGADE C, BARQUET A, MAIBACH HI. Lindane poisonings.
Arch Dermatol. 1983;
119:142
-144
[26] DEVOS S, BOSSCHER KD, STAELS B, BAUER E, ROELS F et coll. Inhibition of cytokine production by the herbicide atrazine; search for nuclear receptor targets.
Biochemical Pharmacology. 2003;
65:303
-308
[27] DITRAGLIA D, BROWN DP, NAMEKATA T, IVERSON N. Mortality study of workers employed at organochlorine pesticide manufacturing plants.
Scand J Work Environ Health. 1981;
7 (suppl 4):140
-146
[28] EL-SHENAWY NS. Effects of insecticides fenitrothion, endosulfan and abamectin on antioxidant parameters of isolated rat hepatocytes.
Toxicol in Vitro. 2010;
24:1148
-1157
[29] EPSTEIN SS. The carcinogenicity of dieldrin. Part I.
Sci Total Environ. 1975;
4:1
-52
[30] EPSTEIN SS. Carcinogenicity of heptachlor and chlordane.
Sci Total Environ. 1976;
6:103
-154
[31] EDWARDS FL, YEDJOU CG, TCHOUNWOU PB. Involvement of oxidative stress in methyl parathion and parathion-induced toxicity and genotoxicity to human liver carcinoma (HepG2) Cells.
Environ Toxicol 2013. 2013 [Epub 2011 May 4];
28:342
-348
[32] FARBER E. Cellular biochemistry of the stepwise development of cancer with chemicals: GHA Clowes memorial lecture.
Cancer Res. 1984;
44:5463
-5474
[33] FAUSTO N, WEBBER EM. Control of liver growth.
Crit Rev Eukaryot Gene Expr. 1993;
3:117
-135
[34] FERNANDEZ V, MASSA L, QUIÒONES L, SIMON-GIAVAROTTI KA, GIAVAROTTI L et coll. Effects of gamma-hexachlorocyclohexane and L-3,3’,5-triiodothyronine on rat liver cytochrome P4502E1-dependent activity and content in relation to microsomal superoxide radical generation.
Biol Res. 2003;
36:359
-365
[35] FOXENBERGA RJ, ELLISONA CA, KNAAKA JB, MAB C, OLSONA JR. CytochromeP450-specific human PBPK/PD models for the organophosphorus pesticides: Chlorpyrifos and parathion.
Toxicology. 2011;
285:57
-66
[36] FROWEIN J. Hypothesis: chemical carcinogenesis mediated by a transiently active carcinogen receptor.
Cytogenet Cell Genet. 2000;
91:102
-104
[37] GALLOWAY SM, ARMSTRONG MJ, REUBEN C, COLMAN S, BROWN B, et coll. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: evaluations of 108 chemicals.
Environ Mol Mutagen. 1987;
10 (suppl 10):1
-175
[38] GANDINI S, AUTIER P, BONIOL M. Reviews on sun exposure and artificial light and melanoma.
Prog Biophys Mol Biol. 2011;
107:362
-366
[39] GOMES-CARNEIRO MR, RIBEIRO-PINTO LF, PAUMGARTTEN FJ. Environmental risk factors for gastric cancer: the toxicologist’s standpoint.
Cad Saude Publica. 1997;
13 (suppl 1) :27
-38
[40] GRASL-KRAUPP B, BURSCH W, RUTTKAY-NEDECKY B, WAGNER A, LAUER B, SCHULTE- HERMANN R. Food restriction eliminates preneoplastic cells through apoptosis and antagonizes carcinogenesis in rat liver.
Proc Natl Acad Sci USA. 1994;
91:9995
-9999
[41] GUZELIAN PS. The clinical toxicology of chlordecone as an example of toxicological risk assessment for man.
Toxicol Lett. 1992;
64-65:589
-596
[42] HAMMOND B, KATZENELLENBOGEN BS, KRAUTHAMMER N, MCCONNELL J. Estrogenic activity of the insecticide chlordécone (Kepone) and interaction with uterine estrogen receptors.
Proc Natl Acad Sci USA. 1979;
76:6641
-6645
[43] HARRIS CJ, WILLIFORD EA, KEMBERLING SR, MORGAN DP. Pesticide intoxications in Arizona.
Ariz Med. 1969;
26:872
-876
[44] HASHIZUME T, YOSHITOMI S, ASAHI S, UEMATSU R, MATSUMURA S et coll. Advantages of human hepatocyte-derived transformants expressing a series of human cytochrome p450 isoforms for genotoxicity examination.
Toxicol Sci. 2010;
116:488
-497
[45] HEIDELBERGER C. Chemical carcinogenesis.
Cancer. 1977;
40:430
-433
[46] HONG HL, BOORMAN GA. Residual myelotoxicity of lindane in mice.
Fundam Appl Toxicol. 1993;
21:500
-507
[47] HUBER MA, KRAUT N, BEUG H. Molecular requirements for epithelial-mesenchymal transition during tumor progression.
Curr Opin Cell Biol. 2005;
17:548
-558
[48] HUFF J. Animal and human carcinogens.
Environ Health Perspect. 1999;
107:A341
-342
[49] HUTTER CM, CHANG-CLAUDE J, SLATTERY ML, PFLUGEISEN BM, LIN Y et coll. Characterization of gene-environment interactions for colorectal cancer susceptibility loci.
Cancer Res. 2012;
72:2036
-2044
[50] IKEGUCHI M, HIROOKA Y, KAIBARA N. Quantitative analysis of apoptosis-related gene expression in hepatocellular carcinoma.
Cancer. 2002;
95:1938
-1945
[51]INVS. DIEYE M, QUENEL P, GORIA S, BLATEAU A, COLONNA M, AZALOUX H. Étude de la répartition spatiale des cancers possiblement liés à la pollution des sols par les pesticides organochlorés, en Martinique, 2009.
[52] JUNQUEIRA V, SIMIZU K, VAN HALSEMA L, KOCH O, BARROS S, VIDELA L. Lindane-induced oxidative stress. I. Time course of changes in hepatic microsomal parameters, antioxidant enzymes, lipid peroxidative indices and morphological characteristics.
Xenobiotica. 1988;
18:1297
-1304
[53] KALENDER S, UZUN FG, DURAK D, DEMIR F, KALENDER Y. Malathion-induced hepatotoxicity in rats: the effects of vitamins C and E.
Food Chem Toxicol. 2010;
48:633
-638
[54] KALLURI R, NEILSON EG. Epithelial-mesenchymal transition and its implications for fibrosis.
J Clin Invest. 2003;
112:1776
-1784
[55] KAMENDULIS LM, KOLAJA KL, STEVENSON DE, WALBORG EF, KLAUNIG JE. Comparative effects of dieldrin on hepatic ploidy, cell proliferation, and apoptosis in rodent liver.
J Toxicol Environ Health. A 2001;
62:127
-141
[56] KISO S, KAWATA S, TAMURA S, ITO N, TAKAISHI K, et coll. Alteration in growth regulation of hepatocytes in primary culture obtained from cirrhotic rat: poor response to transforming growth factor-beta 1 and interferons.
Hepatology. 1994;
20:1303
-1308
[57] KLAUNIG JE, KAMENDULIS LM, XU Y. Epigenetic mechanisms of chemical carcinogenesis.
Hum Exp Toxicol . 2000;
19:543
-555
[58] KLIMISCH HJ, ANDREAE E, TILLMANN U. A systematic approach for evaluating the quality of experimental and ecotoxicological data.
Reg Tox Pharm. 1997;
25:1
-5
[59] KOCAREK TA, SCHUETZ EG, GUZELIAN PS. Regulation of cytochrome P450 2B1/2 mRNAs by Kepone (chlordecone) and potent estrogens in primary cultures of adult rat hepatocytes on Matrigel.
Toxicol Lett. 1994;
71:183
-196
[60] KOLAJA K, STEVENSON D, JOHNSON J, WALBORG E, KLAUNIG JE. Subchronic effects of dieldrin and phenobarbital on hepatic DNA synthesis in mice and rats.
Fundam Appl Toxicol. 1996;
29:219
-228
[61] LARSON PS, EGLE JL, HENNIGAR GR, LANE RW, BORZELLECA JF. Acute, subchronic, and chronic toxicity of chlordecone.
Toxicol Appl Pharmacol. 1979;
48:29
-41
[62] LAUFFENBURGER D, HORWITZ A. Cell migration: a physically integrated molecular process.
Cell. 1996;
84:359
-369
[63] LEE JM, DEDHAR S, KALLURI R, THOMPSON EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease.
J Cell Biol. 2006;
172:973
-981
[64] LEMAIRE G, DE SOUSA G, RAHMANI R A. PXR reporter gene assay in a stable cell culture system: CYP3A4 and CYP2B6 induction by pesticides.
Biochem Pharmacol. 2004a;
68:2347
-2358
[65] LEMAIRE G, TEROUANNE B, MAUVAIS P, MICHEL S, RAHMANI R. Effect of organochlorine pesticides on human androgen receptor activation in vitro.
Toxicol Appl Pharmacol. 2004b;
196:235
-246
[66] LEMAIRE G, MNIF W, PASCUSSI JM, PILLON A, RABENOELINA F, et coll. Identification of new human pregnane X receptor ligands among pesticides using a stable reporter cell system.
Toxicol Sci. 2006;
91:501
-509
[67] LESSEUR C, GILBERT-DIAMOND D, ANDREW AS, EKSTROM RM, LI Z et coll. A case-control study of polymorphisms in xenobiotic and arsenic metabolism genes and arsenic-related bladder cancer in New Hampshire.
Toxicol Lett. 2012;
210:100
-106
[68] LI D, LIU J, LI J. Genotoxic evaluation of the insecticide endosulfan based on the induced GADD153-GFP reporter gene expression.
Environ Monit Assess. 2011;
176:251
-258
[69] LIU Y, WANG H, LIN T, WEI Q, ZHI Y, et coll. Interactions between cigarette smoking and XPC-PAT genetic polymorphism enhance bladder cancer risk.
Oncol Rep. 2012;
28:337
-345
[70] LOOSLI R. Epidemiology of atrazine.
Rev Environ Contam Toxicol. 1995;
143:47
-57
[71] LUCH A. Nature and nurture-lessons from chemical carcinogenesis.
Nat Rev Cancer. 2005;
5:113
-125
[72] MAJUMDAR SK, KOPELMAN HA, SCHNITMAN MJ. Dieldrin-induced chromosome damage in mouse bone-marrow and WI-38 human lung cells.
J Hered. 1976;
67(5):303
-307
[73] MCDONALD JC, MCDONALD AD. The epidemiology of mesothelioma in historical context.
Eur Respir J. 1996;
9:1932
-1942
[74] MEIERHENRY E, RUEBNER B, GERSHWIN ME, HSIEH LS, FRENCH SW. Dieldrin-induced mallory bodies in hepatic tumors of mice of different strains.
Hepatology. 1983;
3:90
-95
[75] MENCOBONI M, LERZA R, BOGLIOLO G, FLEGO G, PANNACCIULLI I. Effect of atrazine on hemopoietic system.
In Vivo. 1992;
6:41
-44
[76] MICHALOPOULOS GK, DEFRANCES MC. Liver regeneration.
Science. 1997;
276:60
-66
[77] MILIGI L, COSTANTINI AS, VERALDI A, BENVENUTI A, VINEIS P, WILL. Cancer and pesticides: an overview and some results of the Italian multicenter case-control study on hematolymphopoietic malignancies.
Ann N Y Acad Sci. 2006;
1076:366
-377
[78] MOLLER S, BECKER U, GRONBAEK M, JUUL A, WINKLER K, SKAKKEBAEK NE. Short-term effect of recombinant human growth hormone in patients with alcoholic cirrhosis.
J Hepatol. 1994;
21:710
-717
[79] MOORE PD, YEDJOU CG, TCHOUNWOU PB, . Malathion-induced oxidative stress, cytotoxicity, and genotoxicity in human liver carcinoma (HepG2) cells.
Environ Toxicol. 2010;
25:221
-226
[80] MOR F, OZMEN O. Effect of vitamin C in reducing the toxicity of endosulfan in liver in rabbits.
Exp Toxicol Pathol. 2010;
62:75
-80
[81] MORTELMANS K, HAWORTH S, LAWLOR T, SPECK W, TAINER B, ZEIGER E. Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals. 1998.
Environ Mutagen. 1986;
8 (suppl 7):1
-119
[82] MULTIGNER L, CORDIER S, KADHEL P, HUC-TERKI F, BLANCHET P, et coll. Pollution par le chlordécone aux Antilles. Quel impact sur la santé de la population ?.
Environnement, Risque et Santé. 2007;
6:405
-407
[83] MULTIGNER L, NDONG JR, GIUSTI A, ROMANA M, DELACROIX-MAILLARD H et coll. Chlordecone exposure and risk of prostate cancer.
J Clin Oncol. 2010;
28:3457
-3462
[84] MUNK ZM, NANTEL A. Acute lindane poisoning with development of muscle necrosis.
Can Med Assoc J. 1977;
117:1050
-1054
[85]NATIONAL CANCER INSTITUTE (NCI).Report on carcinogenesis bioessay of technical grade chlordécone (Kepone) (CAS N° 143-50-0) NTP TR-00. 1976;
[
http://ntp.niehs.nih.gov/].
[86]NATIONAL CANCER INSTITUTE (NCI).Bioassays of aldrin and dieldrin for possible carcinogenicity. National Institute of Health, Division of Cancer Cause and Prevention, Carcinogenesis Program, US Department of Health, Education, and Welfare. Bethesda, MD. N°78-821. 1978;
[87]NATIONAL TOXICOLOGY PROGRAM (NTP).Toxicology and carcinogenesis studies of mirex (CAS No. 2385-85-5) in F344/N rats (feed studies). Research Triangle Park, NC: US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Toxicology Program. NTP TR 3 13, 1990.
[88] NIMS RW, LUBET RA. Induction of cytochrome P-450 in the Norway rat, Rattus norvegicus, following exposure to potential environmental contaminants.
J Toxicol Environ Health. 1995;
46:271
-292
[89] OLIVEIRA PA, COLAÁO A, CHAVES R, GUEDES-PINTO H, DE-LA-CRUZ P, LOPES C. Chemical carcinogenesis.
An Acad Bras Cienc. 2007;
79:593
-616
[90] OLIVER JD, ROBERTS RA. Receptor-mediated hepatocarcinogenesis: role of hepatocyte proliferation and apoptosis.
Pharmacol Toxicol. 2002;
91:1
-7
[91] OMURTAG GZ, TOZAN A, SEHIRLI AO, SENER G. Melatonin protects against endosulfan-induced oxidative tissue damage in rats.
J Pineal Res. 2008;
44:432
-438
[92] ORTON F, ROSIVATZ E, SCHOLZE M, KORTENKAMP A. Widely used pesticides with previously unknown endocrine activity revealed as in vitro antiandrogens.
Environ Health Perspect. 2011;
119:794
-800
[93] PALMITER RD, MULVIHILL ER. Estrogenic activity of the insecticide kepone on the chicken oviduct.
Science. 1978;
201:356
-358
[94] PAOLINI M, BARILLARI J, TRESPIDI S, VALGIMIGLI L, PEDULLI GF, CANTELLI-FORTI G. Captan impairs CYP-catalyzed drug metabolism in the mouse.
Chem Biol Interact. 1999;
123:149
-170
[95] PINCHUK LM, LEE SR, FILIPOV NM. In vitro atrazine exposure affects the phenotypic and functional maturation of dendritic cells.
Toxicol Appl Pharmacol. 2007;
223:206
-217
[96] PITOT HC, DRAGAN YP. Facts and theories concerning the mechanisms of carcinogenesis.
Faseb J. 1991;
5:2280
-2286
[97] REUBER M. Significance of acute and chronic renal disease in Osborne-Mendel rats ingesting dieldrin or aldrin.
Clin Toxicol. 1980;
17:159
-170
[98] REUBER MD. The role of toxicity in the carcinogenicity of endosulfan.
Sci Total Environ. 1981;
20:23
-47
[99] RICHARDSON FC, BOUCHERON JA, DYROFF MC, POPP JA, SWENBERG JA. Biochemical and morphologic studies of heterogeneous lobe responses in hepatocarcinogenesis.
Carcinogenesis. 1986;
7:247
-251
[100] ROSS J, PLUMMER SM, RODE A, SCHEER N, BOWER CC, et coll. Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogens phenobarbital and chlordane in vivo.
Toxicol Sci. 2010;
116:452
-466
[101] RÖCKEN C, CARL-MCGRATH S. Pathology and pathogenesis of hepatocellular carcinoma.
Dig Dis. 2001;
19:269
-278
[102] SAVAGNER P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition.
Bioessays. 2001;
23:912
-923
[103] SHOOK D, KELLER R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development.
Mech Dev. 2003;
120:1351
-1383
[104] SIDDIQUI MK, SAXENA MC, BHARGAVA AK, SETH TD, MURTI CR, KUTTY D. Agrochemicals in the maternal blood, milk, and cord blood: a source of toxicants for prenates and neonates.
Environ Res. 1981;
24:24
-32
[105] SILVA MH, BEAUVAIS SL. Human health risk assessment of endosulfan. I: Toxicology and hazard identification.
Regul Toxicol Pharmacol. 2010;
56:4
-17
[106] SIRICA AE, WILKERSON CS, WU LL, FITZGERALD R, BLANKE RV, GUZELIAN PS. Evaluation of chlordecone in a two-stage model of hepatocarcinogenesis: a significant sex difference in the hepatocellular carcinoma incidence.
Carcinogenesis. 1989;
10:1047
-1054
[107] STARCEVIC SL, BORTOLIN S, WOODCROFT KJ, NOVAK RF. Kepone (chlordecone) disrupts adherens junctions in human breast epithelial cells cultured on matrigel.
In Vivo. 2001;
15:289
-294
[108] STARR HG, CLIFFORD NJ. Acute lindane intoxication: a case study.
Arch Environ Health. 1972;
25:374
-375
[109] STEVENSON DE, WALBORG EF, NORTH DW, SIELKEN RL, ROSS CE, et coll. Monograph: reassessment of human cancer risk of aldrin/dieldrin.
Toxicol Lett. 1999;
109:123
-186
[110] STOREN G. A case of fatal poisoning by jacutin, insecticide and moth protective agent.
Nord Hyg Tidskr. 1955;
36:77
-81
[111] SUNDER RAM RAO CV, SHREENIVAS R, SINGH V, PEREZ-ATAYDE A, WOOLF A. Disseminated intravascular coagulation in a case of fatal lindane poisoning.
Vet Hum Toxicol. 1988;
30:132
-134
[112] TAKEHARA T, LIU X, FUJIMOTO J, FRIEDMAN SL, TAKAHASHI H. Expression and role of Bcl-xL in human hepatocellular carcinomas.
Hepatology. 2001;
34:55
-61
[113] TELANG S, TONG C, WILLIAMS GM. Epigenetic membrane effects of a possible tumor promoting type on cultured liver cells by the non-genotoxic organochlorine pesticides chlordane and heptachlor.
Carcinogenesis. 1982;
3:1175
-1178
[114] TENNEKES H, ELDER L, KUNZ H. Dose-response analysis of the enhancement of liver tumor formation in CF-1 mice by dieldrin.
Carcinogenesis. 1982;
3:941
-945
[115] THIERY JP. Epithelial-mesenchymal transitions in development and pathologies.
Curr Opin Cell Biol. 2003;
15:740
-746
[116] THIERY JP, SLEEMAN JP. Complex networks orchestrate epithelial-mesenchymal transitions.
Nat Rev Mol Cell Biol. 2006;
7:131
-142
[117] UNALDI CORAL N, UCMAN S, YILDIZ H, OZTAS H, DALKILIC S. Potential neoplastic effects of parathion-methyl on rat liver.
J Environ Sci. 2009;
21:696
-699
[118]US EPA.Summary of reported pesticide incidents involving chlordane. Pesticide incident monotoring system. US. Environmental Protection Agency, Office of pesticide programs. Washington DC, US. Report, n° 360 [
http://www.epa.gov/epahome/search.html]. 1980;
[119]US EPA.Health Effects Support Document for Aldrin/Dieldrin. 2003a;
[120]US EPA. Toxicological Profile for Atrazine.
2003b;
[122]US EPA.NRDC comments on Captan RED, OPP-2004-0296, Jan 24. 2005;
[123]US EPA.IRIS Toxicological Review and Summary Documents for Chlordecone (Kepone) (2009 Final). US Environmental Protection Agency, Washington, DC. 2009;
[124]US EPA.Re-Evaluation of Human Health Effects of Atrazine: Review of Non-Cancer Epidemiology, Experimental Animal and In vitro Studies. 2010;
[125]US NAS. An evaluation of the carcinogenicity of chlordane and heptachlor.
Natl Acad Sci J. Washington, DC, US. 1977;
[126] VIDELA LA, ARISI AC, FUZARO AP, KOCH OR, JUNQUEIRA VB. Prolonged phenobarbital pretreatment abolishes the early oxidative stress component induced in the liver by acute lindane intoxication.
Toxicol Lett. 2000;
115:45
-51
[127] VIDELA LA, BARROS SB, JUNQUEIRA VB. Lindane-induced liver oxidative stress.
Free Radic Biol Med. 1990;
9:169
-179
[128] WEICHENTHAL S, MOASE C, CHAN P. A review of pesticide exposure and cancer incidence in the Agricultural Health Study cohort.
Environ Health Perspect. 2010;
118:1117
-1125
[129] WETZEL LT, LUEMPERT LG, BRECKENRIDGE CB, TISDEL MO, STEVENS JT, et coll. Chronic effects of atrazine on estrus and mammary tumor formation in female Sprague-Dawley and Fischer 344 rats.
J Toxicol Environ Health. 1994;
43:169
-182
[130] WILLIAMS GM. Mechanisms of chemical carcinogenesis and application to human cancer risk assessment.
Toxicology. 2001;
166:3
-10
[131] WILSON VS, LEBLANC GA. Endosulfan elevates testosterone biotransformation and clearance in CD-1 mice.
Toxicol Appl Pharmacol. 1998;
148:158
-168
[132] XU J, YIN Z, HUANG M, WANG X, GAO W, et coll. Polymorphisms and Cancer risk: a Meta-analysis Based on 33 Case-control Studies.
Asian Pac J Cancer Prev. 2012;
13:901
-907
[133] XUE Y, WANG M, ZHONG D, TONG N, CHU H, SHENG X, ZHANG Z. ADH1C Ile350Val Polymorphism and Cancer Risk : Evidence from 35 Case-Control Studies.
PLoS One. 2012;
7:e37227
[134] YUSPA SH, HENNINGS H, LICHTI U, KULESZ-MARTIN M. Organ specificity and tumor promotion.
Basic Life Sci. 1983;
24:157
-171
[135] ZUCCHINI N, DE SOUSA G, BAILLY-MAITRE B, GUGENHEIM J, BARS R, et coll. Regulation of Bcl-2 and Bcl-xL anti-apoptotic protein expression by nuclear receptor PXR in primary cultures of human and rat hepatocytes.
Biochim Biophys Acta. 2005;
1745:48
-58
[136] ZUCCHINI-PASCAL N, DE SOUSA G, RAHMANI R. Lindane and cell death: at the crossroads between apoptosis, necrosis and autophagy.
Toxicology. 2009;
256:32
-41
[137] ZUCCHINI-PASCAL N, DE SOUSA G, PIZZOL J, RAHMANI R. Molecular investigation of the effects of lindane in rat hepatocytes: Microarray and mechanistic studies.
Food Chem Toxicol. 2011;
49:3128
-3135
→ Aller vers SYNTHESE