
Un nombre croissant de données cliniques et expérimentales confirme le rôle important joué par le stress oxydant dans la pathogénie et la progression des maladies cardiovasculaires, particulièrement l’athérosclérose et l’hypertension [ 1]. Par exemple, une corrélation directe a été établie entre le développement de l’hypertension et la proportion d’anions superoxydes (
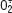 ) dans les vaisseaux des rats spontanément hypertendus (SHR) [
2].
) dans les vaisseaux des rats spontanément hypertendus (SHR) [
2].
Des études récentes ont établi que l’aspirine (AAS) est un agent antioxydant puissant qui réduit l’activité de la NAD(P)H oxydase et la production de l’
dans le cœur et les vaisseaux. Le rétablissement de l’ à des valeurs normales par le traitement de rats SHR à l’AAS a été accompagné d’une atténuation de la hausse de la tension artérielle (TA) et d’une amélioration des fonctions endothéliales de la vasodilatation chez ces animaux [
3]. Le traitement à l’AAS a aussi prévenu le stress oxydant, l’hypertension artérielle et l’hypertrophie cardiaque provoquées, chez le rat, par l’administration chronique d’angiotensine II (Ang II) [
4], un agent causal majeur de la pathogénie de plusieurs maladies cardiovasculaires : athérosclérose, hypertension, insuffisance cardiaque et remodelage ou hypertrophie cardiaque [
5]. Plusieurs études suggèrent donc que l’activation de la NAD(P)H oxydase et l’augmentation de la production de l’, qui y est associée, constituent l’un des mécanismes majeurs qui sous-tendent les effets délétères de l’activation des récepteurs AT1 sur le système cardiovasculaire. De plus, il a été proposé que l’Ang II contribue à augmenter, entre autres, la surexpression de la COX-2 dans les tissus cardiovasculaires. Nous avons donc cherché à évaluer les rôles respectifs des cyclo-oxygénases de type 1 et 2 (COX-1 et 2) pour contrer les effets inflammatoires et pro-oxydants de l’Ang II [
6].
Les deux isoformes des COX convertissent l’acide arachidonique en différents types de prostaglandines et de thromboxanes. La COX-1 est exprimée de façon ubiquitaire dans la plupart des tissus de mammifère, principalement dans l’estomac, les plaquettes et les vaisseaux sanguins. L’activité de la COX-1 est constitutive des prostanoïdes dont elle assure la production de façon relativement stable. À l’opposé, la COX-2 est indétectable dans la plupart des tissus, mais peut être induite par plusieurs cytokines dans les macrophages, le cerveau, les chondrocytes, les fibroblastes et les cellules synoviales.
Les agents anti-inflammatoires non-stéroïdiens inhibent l’activité des COX et sont divisés en trois classes, l’AAS (inhibiteur irréversible des COX-1 et COX-2) les inhibiteurs sélectifs de COX-2 (par exemple, rofécoxib) et les inhibiteurs non sélectifs de COX (par exemple, ibuprofène) [ 7]. Il n’existe aucune donnée montrant une relation causale entre l’activité des COX et le degré de stress oxydant, mais des études suggèrent que la COX-2 intervient dans l’amorce de la production de radicaux libres [ 8].
Une perfusion chronique d’Ang II (200 ng/kg/min) par des pompes osmotiques implantées sous la peau des rats augmente l’activité de la NAD(P)H oxydase et la production de l’
dans les tissus cardiovasculaires [6]. Parallèlement à cette augmentation progressive de l’, la TA s’élève graduellement et simultanément, l’administration chronique de l’Ang II augmente l’expression cardiaque de la COX-2 sans affecter l’expression de la COX-1 [6]. Nous avons observé que le traitement simultané avec l’AAS a normalisé l’expression de COX-2, la production de l’ et la TA chez ces animaux. Des effets similaires à ceux de l’AAS ont été observés après un traitement avec le rofécoxib. En effet, l’AAS exerce son action anti-Ang II par inhibition de l’expression des COX-2 et du stress oxydant qui est associé à l’activation de cette enzyme. En revanche, des inhibiteurs non sélectifs des COX (ibuprofène, indométacine) n’ont pas eu d’effets antioxydants ou préventifs de l’élévation de la TA [6]. De plus, les inhibiteurs non sélectifs n’ont pu bloquer l’expression des COX2 induite par l’Ang II. Ces résultats suggèrent donc que les COX-2 exerceraient un rôle proéminent dans l’induction du stress oxydant et dans l’effet hypertenseur de l’Ang II. Nos observations sont en accord avec celles d’une autre étude qui a révélé une augmentation de l’expression de la COX-2 par l’Ang II dans les cellules de muscles lisses vasculaires [
9].
L’AAS et le rofécoxib ont protégé les tissus contre les effets de l’Ang II par leurs effets inhibiteurs sur la voie COX-2, ainsi que par leurs effets antioxydants plutôt que par leurs effets anti-inflammatoires, puisque les anti-inflammatoires non sélectifs n’ont pas prévenu l’augmentation de l’
ou l’élévation de la TA chez ces animaux. Nos résultats démontrent donc une relation étroite entre l’activation de la voie COX-2 et la production locale de l’ dans les tissus cardiovasculaires lors du traitement chronique à l’Ang II. L’inhibition des COX-2 semble donc constituer une option thérapeutique potentiellement bénéfique pour le traitement de maladies liées à une augmentation du stress oxydant par une hyperactivité du système rénine-angiotensine (Figure 1). Bien qu’agissant de préférence sur la voie COX-1, l’aspirine affecte aussi la voie COX-2.
| Figure 1.
Schéma des effets inhibiteurs par l’aspirine et le rofécoxib sur des actions vasculaires de l’Ang II. L’angiotensine II induit un stress oxydant dans les tissus cardiovasculaires par l’augmentation de l’activité de la NAD(P)H oxydase et par la production de radicaux libres. L’expression et l’activité de la cyclo-oxygénase-2 (COX-2) dans les tissus cardiovasculaires sont aussi stimulées par l’Ang II. Le stress oxydatif et l’activation de COX-2, ainsi que l’activation du facteur nucléaire kappa B (NF-κB) et l’augmentation de la perméabilité des vaisseaux par l’Ang II favorisent l’inflammation locale. Tous ces facteurs contribuent plus ou moins à la pathogénie et à la progression des maladies cardiovasculaires. L’AAS et le rofécoxib protègent les tissus cardiovasculaires contre l’Ang II par l’inhibition de la production de l’O2
- ainsi que par la diminution de l’expression et de l’activité de COX-2. |

La signification clinique de nos observations ne peut être que spéculative. L’AAS semble posséder des propriétés antioxydantes importantes qui pourraient jouer un rôle dans la protection du système cardiovasculaire chez l’homme. Par ailleurs, les effets antihypertenseurs et antidiabétiques de l’AAS pourraient également jouer un rôle important en prévenant le développement du diabète de type 2 et en atténuant la progression de l’hypertension artérielle. L’étude récente du groupe de Hermida et al. [ 10] est particulièrement prometteuse à cet égard. En effet, ces chercheurs ont démontré qu’un traitement de trois mois avec de l’AAS à raison de 100 mg pris au coucher provoquait une chute de la TA de 6,8/4,8 mmHg chez des patients hypertendus de grade I non traités. En revanche, l’AAS administrée le matin a eu pour effet d’augmenter significativement la TA de 2,6/1,6 mmHg, suggérant l’existence d’une fenêtre chronobiologique précise pour l’effet antihypertenseur de l’AAS. Ces nouvelles observations soulèvent donc de nouvelles interrogations dont les réponses contribueront à long terme à mieux connaître les mécanismes de régulation de la TA.
