| |
| Med Sci (Paris). 2006 January; 22(1): 47–53. Published online 2006 January 15. doi: 10.1051/medsci/200622147.Les espèces actives de l’oxygène : le yin et le yang de la mitochondrie Audrey Carrière,1 Anne Galinier,1,2 Yvette Fernandez,1 Maria-Carmen Carmona,1 Luc Pénicaud,1 and Louis Casteilla1* 1UMR 5018 CNRS-UPS, IFR 31, CHU Rangueil, TSA 50032, 31059 Toulouse Cedex 9, France 2Groupe de recherche et d’étude en nutrition (GREEN), Laboratoire de biochimie générale et nutritionnelle, place du Docteur Baylac, CHU Purpan, TSA 40031, 31059 Toulouse Cedex 9, France |
Effets cellulaires des EAO : pourquoi tant de controverses bibliographiques ? Des termes à définir Les espèces actives de l’oxygène (EAO) sont des radicaux libres ou des molécules [
1]. Un radical libre est une espèce chimique, neutre ou chargée, qui a la particularité de porter un électron célibataire (ou non apparié) sur sa couche externe, ce qui le rend généralement instable et capable de réagir plus ou moins rapidement avec d’autres molécules chimiques environnantes. Les réactions de transfert d’électrons qu’il produit (réactions d’oxydoréduction, redox) conduisent souvent à la formation d’un nouveau radical, ce phénomène pouvant se propager par des réactions en chaîne. Les réductions mono-électroniques successives de l’oxygène donnent naissance à différentes EAO : l’anion superoxyde (
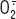 ), le peroxyde d’hydrogène (H2O2) et le radical hydroxyle (.OH). Au sein de la cellule, tous les processus utilisant de l’oxygène dans les différents compartiments subcellulaires sont capables de produire des EAO. ), le peroxyde d’hydrogène (H2O2) et le radical hydroxyle (.OH). Au sein de la cellule, tous les processus utilisant de l’oxygène dans les différents compartiments subcellulaires sont capables de produire des EAO. Afin de limiter la concentration en EAO, les cellules sont équipées de divers systèmes anti-oxydants qui agissent de plusieurs manières [1] : en inhibant la formation des EAO (la séquestration des métaux de transition empêche la réaction de Fenton et la production de .OH), en les métabolisant grâce à des enzymes (superoxyde dismutase, catalase et glutathion peroxydase) ou à des piégeurs de radicaux libres (glutathion, vitamine C, vitamine E, coenzyme Q) ou, encore, en réparant les dommages oxydatifs (par des molécules, glutathion et thiorédoxine réduite, capables de réduire les résidus cystéine préalablement oxydés par les EAO). Les effets des EAO en biologie cellulaire sont le sujet de nombreuses controverses bibliographiques [
2]. À titre d’exemple, alors que les EAO peuvent dans certaines situations stimuler la prolifération cellulaire et être ainsi impliquées dans la transformation oncogénique et le cancer, elles sont également considérées comme des acteurs fondamentaux régulant les processus d’apoptose et induisant la mort cellulaire [2]. Une des explications à cet apparent paradoxe réside dans le fait que les réponses cellulaires varient de manière très sensible et très différente en fonction de la nature chimique, du niveau de production ou encore du véritable site subcellulaire producteur d’EAO. Nature chimique, concentration et sources cellulaires des EAO : trois paramètres, une grande complexité L’importance de la nature chimique des EAO est parfois sous-estimée dans les recherches, au profit de l’utilisation d’un système producteur d’EAO (ou un agent oxydant) donné, sensé représenter une source d’EAO « standard » : il est pourtant décrit depuis très longtemps que
, H2O2 et .OH ont des demi-vies et des réactivités très différentes [
3]. Une étude récente chez la levure montre bien que les réponses cellulaires dépendent spécifiquement de la nature de l’agent oxydant utilisé [
4] : parmi 456 mutants de levure, seuls 2 sont sensibles (croissance altérée) à l’ensemble des 5 agents oxydants utilisés. Cela signifie que des oxydants différents mettent en jeu des fonctions cellulaires spécifiques à chacun et engendrent des réponses cellulaires différentes, soulignant la grande complexité du système redox à l’échelle cellulaire. Un des moyens permettant de démontrer l’implication spécifique d’une EAO donnée sur une fonction cellulaire donnée est d’utiliser des piégeurs spécifiques de chaque EAO, ou de surexprimer les enzymes impliquées dans leur métabolisme. La concentration en EAO, qui résulte de l’équilibre entre les systèmes producteurs et les systèmes anti-oxydants, détermine également leurs effets cellulaires. Une production relativement modérée d’EAO associée à des systèmes de gestion efficaces peut induire une augmentation modérée, aiguë et transitoire de leur concentration (Figure 1), les EAO pouvant alors modifier de manière réversible certaines molécules environnantes : ainsi, l’oxydation de résidus cystéine de protéines peut induire une modification de leur activité [
5]. C’est ainsi que la production d’EAO, après fixation de l’insuline sur son récepteur, inactive la protéine tyrosine phosphatase-1B (PTP-1B), ce qui augmente la phosphorylation des résidus tyrosine du récepteur de l’insuline et stimule la voie de signalisation en aval [
6] : dans ces conditions, les EAO agissent comme de véritables seconds messagers. En revanche, un excès de production d’EAO ou une diminution de l’activité des systèmes anti-oxydants conduisent à la mise en place d’un stress oxydant (Figure 1) : les modifications des molécules environnantes sont alors non spécifiques et irréversibles, ce qui entraîne une stimulation chronique des voies de signalisation [2]. C’est dans ce contexte que les EAO deviennent toxiques et sont impliquées dans le vieillissement et certaines pathologies.  | Figure 1.
Conséquences cellulaires des EAO : une question d’équilibre. Si la production d’EAO est modérée et relativement courte dans le temps, un déséquilibre temporaire dans la balance entre production d’EAO et défenses anti-oxydantes est observé : les EAO activent de manière spécifique et réversible des voies de signalisation qui permettront notamment de déclencher des systèmes d’adaptation de la cellule. En cas de production d’EAO trop intense et maintenue dans le temps, les systèmes anti-oxydants sont dépassés, et un dérèglement chronique de l’équilibre provoque l’établissement d’un stress oxydant aboutissant à la survenue de situations physiopathologiques [ 2]. |
Par ailleurs, l’existence de nombreuses sources de production d’EAO dans la cellule ajoute un niveau supplémentaire de complexité. Une production extracellulaire d’EAO par la NADPH oxydase doit avoir des répercussions cellulaires très différentes de celle provenant de la membrane interne de la mitochondrie, ne serait-ce que par la présence de systèmes de détoxification différents dans ces divers sites subcellulaires. |
Production mitochondriale d’EAO : une production à part, reflet de l’activité cellulaire La mitochondrie représente le site majeur de production cellulaire d’EAO : dans les cellules non phagocytaires, 80 % de l’anion superoxyde proviennent du fonctionnement de la chaîne respiratoire. Ces EAO mitochondriales, dont la production est dépendante du fonctionnement de la chaîne respiratoire, sont spécifiquement considérées comme des senseurs de l’environnement, permettant aux cellules de s’adapter à ses fluctuations. C’est pour ces raisons que nous avons limité cet article aux EAO d’origine mitochondriale, bien que cette production ne recouvre pas toute la réalité de la réponse cellulaire. La production des EAO mitochondriales est intrinsèque au fonctionnement de la chaîne respiratoire Les voies métaboliques associées aux différents nutriments convergent vers l’acétylCoA, dont la prise en charge par le cycle de Krebs libère de l’énergie sous forme d’équivalents réduits, NADH,H + et FADH 2, donneurs d’électrons à la chaîne respiratoire (Figure 2). Les électrons sont transférés le long de la chaîne respiratoire au cours de réactions d’oxydoréduction jusqu’à l’accepteur final, l’oxygène, qui est réduit complètement en H 2O. L’énergie ainsi libérée (sous forme d’un gradient de protons de part et d’autre de la membrane interne) est alors utilisée par l’ATP synthétase pour synthétiser l’ATP : ce sont les oxydations phosphorylantes [
7]. Dans ces conditions, le fonctionnement de l’ATP synthétase est couplé à celui de la chaîne respiratoire.  | Figure 2.
Production mitochondriale et prise en charge de l’anion superoxyde. Au cours du transfert des électrons, dans des conditions physiologiques, il peut y avoir des fuites d’électrons et production d’anion superoxyde (
) par le complexe I et le coenzyme Q. est métabolisé par la superoxyde dismutase (SOD2, ou MnSOD, étant l’isoforme spécifiquement localisée dans la matrice mitochondriale) en peroxyde d’hydrogène (H 2O 2). Celui-ci peut être décomposé en H 2O par la glutathion peroxydase (GPx1), en présence de glutathion réduit (GSH), se transformer en radical hydroxyle ( .OH) par la réaction de Fenton, en présence de Fe 2+, ou encore diffuser dans le cytoplasme et modifier l’activité de protéines cytoplasmiques ou nucléaires en les oxydant. Le radical hydroxyle peut également oxyder les protéines, ou l’ADN (mitochondrial et nucléaire), et induire la peroxydation lipidique (elle-même limitée par des piégeurs de radicaux libres, de nature lipophile, tels que la vitamine E ou le coenzyme Q). Il est important de souligner ici la dualité d’action du coenzyme Q : piégeur de radicaux libres, sous forme réduite, dans les membranes biologiques, il est la source essentielle d’O -.
2 dans la chaîne respiratoire. |
Une proportion significative de l’oxygène (2 % à 6 %) échappe à la réduction complète en H2O et subit une réduction mono-électronique au niveau des complexes I et III de la chaîne respiratoire [
8], pour donner naissance à l’ion superoxyde (
) : cette production est absolument indissociable du processus respiratoire. Plusieurs systèmes anti-oxydants mitochondriaux, localisés en différents sites, prennent en charge et ses dérivés (Figure 2). La comparaison des phénotypes de souris invalidées pour les différentes isoformes de la superoxyde dismutase (SOD) démontre bien les conséquences spécifiquement liées à l’ion mitochondrial : l’invalidation génique de la SOD mitochondriale entraîne en effet une létalité néonatale [
9], alors que les souris invalidées pour les SOD cytosolique ou extracellulaire n’ont aucun problème de viabilité [
10,
11]. Lorsque le fonctionnement de la chaîne respiratoire est normal et que le potentiel de membrane est faible, la production d’
par les complexes I et III s’effectue au cours du transfert des électrons du complexe I au cycle des quinones (forward electron transfer). Au niveau du complexe III, c’est le coenzyme Q, sous sa forme ubisemiquinone QH., qui est responsable de la production d’. Lorsque le potentiel de membrane est élevé (en l’absence d’ADP) et lorsque du FADH2 est utilisé par le complexe II, il y a production d’ au sein du complexe I via le flux inverse (reverse electron transfer) [
12]. Modulation de la production des EAO mitochondriales La production d’
par les complexes I et III est un processus non enzymatique, contrôlé par la loi d’action de masse. Elle est proportionnelle à la concentration en oxygène et à l’état de réduction des transporteurs (ils sont potentiellement donneurs d’électrons et producteurs d’ à l’état réduit). Lors d’un apport excessif de substrats par rapport aux réels besoins énergétiques de la cellule (rapport ATP/ADP élevé), l’arrivée massive d’électrons sans dissipation du potentiel de membrane par l’ATP synthétase peut être à l’origine d’une surproduction d’EAO : il a notamment été démontré que l’hyperglycémie augmente la production d’EAO [
13]. Par ailleurs, certaines biomolécules capables d’inhiber le transfert des électrons au sein du complexe III, le céramide par exemple, augmentent la production d’EAO [
14]. De même, des agents pharmacologiques comme la roténone ou l’antimycine (inhibiteurs des complexes I et III, respectivement) sont utilisés pour moduler la production d’EAO mitochondriales et étudier leurs conséquences cellulaires. À l’inverse, toute situation diminuant de façon modérée le potentiel de membrane a tendance à diminuer la production d’
: c’est la théorie du mild uncoupling [
15]. En effet, lors d’un découplage modéré entre le fonctionnement de la chaîne respiratoire et celui de l’ATP synthétase, les transporteurs sont dans un état plutôt oxydé et la production d’ est faible. Les protéines découplantes UCP (uncoupling protein), insérées dans la membrane interne mitochondriale, sont ainsi capables de moduler la production d’EAO [
16] : cela est conforté par le fait que des animaux génétiquement invalidés pour le gène UCP3 ont des signes de stress oxydant notables [
17]. Les UCP peuvent être intégrées au système de gestion des EAO mitochondriales et sont d’autant plus efficaces qu’elles empêchent la formation d’ [
18]. |
Effets des EAO mitochondriales EAO mitochondriales : éléments clés dans le contrôle physiologique de l’homéostasie énergétique Un étude récente propose que les EAO mitochondriales produites lors d’un excès de substrats énergétiques (comme le pyruvate) déclenchent une voie de signalisation spécifique qui ré-oriente le métabolisme vers une voie de stockage du glucose, plutôt que vers son utilisation oxydative [
19] (Figure 3) : en limitant l’apport d’équivalents réduits à la chaîne respiratoire, et donc la production d’EAO, ce mécanisme constitue un rétrocontrôle négatif effectué par les EAO sur leur propre production. Ces travaux ont ainsi, pour la première fois, mis en évidence le rôle des EAO mitochondriales en tant que senseurs des conditions nutritionnelles, capables de réguler le métabolisme.  | Figure 3.
EAO mitochondriales, médiatrices des multiples conséquences du métabolisme. Une concentration élevée en nutriments entraîne une augmentation du potentiel redox des couples NADH,H+ et FADH2, ce qui produit une entrée massive d’électrons au sein de la chaîne respiratoire (CR). Si les besoins énergétiques réels de la cellule sont faibles (état IV de la respiration, c’est-à-dire peu d’ADP à phosphoryler), l’ATP synthétase ne peut fonctionner ; le gradient de protons ne peut donc être dissipé, et le potentiel de membrane est élevé. Cela ralentit le flux d’électrons au sein de la chaîne respiratoire, et la concentration en transporteurs d’électrons réduits augmente : la production d’
et, en conséquence, celle des autres EAO sont alors élevées. Les EAO mitochondriales ainsi produites sont responsables des conséquences liées à l’intensité du métabolisme : adaptation des voies métaboliques, complications diabétiques ou encore implication dans le vieillissement. |
Étant donné cette étroite relation entre métabolisme et EAO mitochondriales, nous avons suggéré que les EAO mitochondriales pourraient contrôler le développement du tissu adipeux, le principal organe de stockage énergétique de l’organisme. À l’aide d’approches pharmacologiques permettant de moduler finement et de manière modérée leur production, nous avons démontré que les EAO mitochondriales contrôlent deux des processus fondamentaux du développement du tissu adipeux : la prolifération des précurseurs adipocytaires, mais aussi leur différenciation en adipocytes, et ce en activant des voies de signalisation spécifiques (induction du facteur de transcription CHOP-10/GADD153) (C/EBP homologous protein/growth arrest and DNA-damage-inducible protein) [
20,
21]. EAO mitochondriales : médiateurs des effets de l’hypoxie Deux théories concernant l’implication des EAO en hypoxie s’affrontent [
22]. La première est fondée sur le rôle joué par la NADPH oxydase : comme celle-ci produit l’anion superoxyde proportionnellement à la concentration en oxygène, de faibles pressions en oxygène devraient diminuer la production d’EAO. Une deuxième théorie décrit au contraire une augmentation de la production d’EAO en situation d’hypoxie, par l’intermédiaire de la chaîne respiratoire mitochondriale [
23,
24], cette production mitochondriale d’EAO étant le résultat de l’inhibition de l’activité de la cytochrome oxydase [ 8]. La quantification des EAO après hypoxie semble être un des moyens pour valider l’une ou l’autre des hypothèses selon les cellules [ 22]. Les modifications de la production d’EAO pourraient être responsables des conséquences, sur l’activité transcriptionnelle de la cellule, de l’hypoxie. L’activation transcriptionnelle de nombreux gènes en hypoxie est sous la dépendance du facteur de transcription HIF-1 (hypoxia inducible factor) (Figure 4) : en situation d’hypoxie, les EAO mitochondriales seraient responsables de la stabilisation de la sous-unité régulatrice HIF-1α, et donc de l’activation d’HIF-1 ; en effet, celle-ci est bloquée en l’absence de mitochondries fonctionnelles [23, 24]. Ainsi, la production mitochondriale d’EAO permettrait aux cellules de s’adapter à cette condition environnementale particulière.  | Figure 4.
Régulation de l’activité d’HIF-1α par les EAO mitochondriales. Le facteur de transcription HIF-1 (hypoxia inducible factor) est composé de deux sous-unités : HIF-1β, connue sous le nom d’ARNT (aryl hydrocarbon nuclear translocator), et HIF-1α, sous-unité régulatrice. En normoxie, HIF-1α est dégradée par le système ubiquitine (Ub)-protéasome après hydroxylation de certains résidus proline par des proline hydroxylases. La modification de la quantité intracellulaire d’EAO en hypoxie (augmentation selon la théorie de la chaîne respiratoire) serait responsable de l’inhibition des proline hydroxylases et donc de l’accumulation de HIF-1α, qui pourrait alors moduler la transcription de gènes activant l’angiogenèse, la glycolyse et l’érythropoïèse, ou inhibant l’adipogenèse. Protéine VHL : protéine von Hippel-Lindau ; VEGF : vascular endothelium growth factor ; Glut1 : transporteur du glucose ; PPAR : récepteur activé par les proliférateurs de peroxysomes ; EPO : érythropoïétine. |
Dans le cadre du tissu adipeux, nous avons montré que l’effet inhibiteur de l’hypoxie sur la différenciation adipocytaire [
25] était en partie dû aux EAO mitochondriales, qui induiraient de manière indépendante deux voies de signalisation anti-adipogéniques : celle d’HIF-1α et celle de CHOP-10/GADD153 [21]. Stress oxydant d’origine mitochondriale : toxicité du glucose et vieillissement Des études ont démontré que la production mitochondriale d’EAO induite par l’hyperglycémie altère le fonctionnement des cellules endothéliales et pourrait être à l’origine des micro-angiopathies observées dans le diabète [
26]. En effet, l’inhibition de la production d’EAO par la surexpression d’UCP1 ou de la SOD mitochondriale, qui prévient les conséquences délétères de l’hyperglycémie sur les cellules, constitue une preuve de l’implication spécifique de la mitochondrie comme site majeur de la production d’
(Figure 3) [13]. Ainsi, un stress oxydant mitochondrial produit par une hyperglycémie chronique peut être directement impliqué dans la toxicité du glucose et les complications diabétiques. Qui plus est, ces expériences sembleraient indiquer que c’est spécifiquement l’anion superoxyde qui serait responsable de la mise en place de ces dysfonctionnements. La mitochondrie est non seulement source d’EAO, mais aussi la cible directe de leurs conséquences délétères, le stress oxydant d’origine mitochondriale étant décrit comme une des causes majeures du vieillissement [
27]. Les fonctions, la morphologie et le renouvellement des mitochondries sont altérés au cours du vieillissement, notamment en raison de l’accumulation de mutations somatiques de l’ADN mitochondrial, une cible privilégiée des EAO produites par la chaîne respiratoire (Figures 1 et 2). Le mauvais fonctionnement des oxydations phosphorylantes dans ces mitochondries « vieillissantes » entraîne une production très élevée d’EAO, accroissant le taux de mutations de l’ADN mitochondrial et donnant ainsi naissance au « cycle vicieux » de la mitochondrie : le mutant mev-1 du nématode C. elegans, déficient pour le complexe II de la chaîne respiratoire, présente ainsi des dommages oxydatifs et une diminution de 30 % de sa durée de vie [
28]. Le traitement de ces vers mev-1 par des composés ayant des activités SOD-et catalase-like restaure une durée de vie égale à celle des vers sauvages, démontrant le rôle fondamental des EAO mitochondriales dans le processus du vieillissement. De plus, et toujours chez C. elegans, l’effet bénéfique de la restriction calorique sur la durée de vie serait dû à une diminution de la production mitochondriale d’EAO associée à une moindre quantité d’équivalents réduits NADH,H+ et FADH2, conséquence de la faible disponibilité en substrats énergétiques (Figure 3) [
29]. Chez l’homme, l’augmentation des dommages oxydatifs au cours de l’hyperglycémie et du vieillissement, mais aussi au cours de certaines maladies mitochondriales (syndromes MELAS [mitochondrial encephalomyopathy, lactic acidosis and stroke like episodes] et MERRF [myoclonic epilepsy and ragged red fibres]) ou neurodégénératives (Parkinson, Alzheimer), pourrait être liée à une surproduction d’EAO mitochondriales. |
Conclusions et perspectives Les EAO mitochondriales, acteurs fondamentaux de l’adaptation et de la survie des cellules face aux fluctuations de l’environnement, peuvent également être à l’origine du vieillissement cellulaire et de nombreuses pathologies, et constituer des éléments limitant la durée de vie des organismes aérobies. Plusieurs stratégies thérapeutiques utilisant des supplémentations en anti-oxydants sont actuellement proposées, avec des résultats plus ou moins satisfaisants. Des molécules (vitamine E, coenzyme Q) liées à des groupements chimiques les dirigeant vers la mitochondrie [
30] pourraient constituer un espoir dans la prévention et le traitement des pathologies impliquant les EAO mitochondriales. ‡ |
Footnotes |
1. Gutteridge JM. Biological origin of free radicals, and mechanisms of antioxidant protection. Chem Biol Interact 1994; 91 : 133–40. 2. Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002; 82 : 47–95. 3. Halliwell B, Gutteridge JMC. Free radicals in biology and medecine. Oxford : Clarendon Press, 1989 : 22–81. 4. Thorpe GW, Fong CS, Alic N, et al. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc Natl Acad Sci USA 2004; 1 : 6564–9. 5. Morel Y, Barouki R. Repression of gene expression by oxidative stress. Biochem J 1999; 342 : 461–96. 6. Mahadev K, Wu X, Zilbering A, et al. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem 2001; 276 : 48662–9. 7. Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature 1967; 213 : 137–9. 8. Turrens J.-F. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552 : 335–44. 9. Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995; 11 : 376–81. 10. Reaume AG, Elliott JL, Hoffman EK, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 1996; 13 : 43–7. 11. Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci USA 1995; 92 : 6264–8. 12. Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 2002; 80 : 780–7. 13. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404 : 787–90. 14. Garcia-Ruiz C, Colell A, Mari M, et al. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem 1997; 272 : 11369–77. 15. Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely lowlevels of oxygen and its one-electron reductants. Q Rev Biophys 1996; 29 : 169–202. 16. Nègre-Salvayre A, Hirtz C, Carrera G, et al. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 1997; 11 : 809–15. 17. Vidal-Puig AJ, Grujic D, Zhang CY, et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem 2000; 275 : 16258–66. 18. Casteilla L, Rigoulet M, Pénicaud L, Casteilla L. Mitochondrial ROS metabolism: modulation by uncoupling proteins. IUMBM Life 2001; 52 : 181–8. 19. Nemoto S, Takeda K, Yu ZX, et al. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol 2000; 20 : 7311–8. 20. Carrière A, Fernandez Y, Rigoulet M, et al. Inhibition of white preadipocyte proliferation by mitochondrial reactive oxygen species. FEBS Lett 2003; 550 : 163–7. 21. Carrière A, Carmona MC, Fernandez Y, et al. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: a mechanism for hypoxia-dependent effect. J Biol Chem 2004; 279 : 40462–9 22. Semenza GL. Perspectives on oxygen sensing. Cell 1999; 98: 281–4. 23. Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275 : 25130–8. 24. Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 2002; 283 : L922–31. 25. Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell 2002; 2 : 331–41. 26. Pfeiffer A, Schatz H. Diabetic microvascular complications and growth factors. Exp Clin Endocrinol Diabetes 1995; 103 : 7–14. 27. Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med 2003; 35 : 1–8. 28. Melov S, Ravenscroft J, Malik S, et al. Extension of life-span with superoxide dismutase/catalase mimetics. Science 2000; 289 : 1 567–9. 29. Lin SJ, Ford E, Haigis M, et al. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev 2004; 18 : 12–6. 30. Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J 2003; 17 : 1972–4. |





