| |
| Med Sci (Paris). 2008 March; 24(3): 277–283. Published online 2008 March 15. doi: 10.1051/medsci/2008243277.Les bases moléculaires de l’approvisionnement en cuivre Leçons tirées de la levure Julie Laliberté and Simon Labbé* Département de Biochimie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, 3001, 12e Avenue Nord, Sherbrooke (Québec), J1H 5N4 Canada |
Le cuivre (Cu) est un oligo-élément essentiel à tous les organismes vivants. Dans de nombreuses réactions enzymatiques, il est courant d’observer les constants allers-retours de cet ion métallique de l’état oxydé (Cu2+) à l’état réduit (Cu1+). Cette propriété du cuivre en fait un excellent cofacteur. Le cuivre loge au cœur du site catalytique de beaucoup d’enzymes essentiels pour les cellules. Des activités cellulaires comme la respiration, le transport du fer et la protection contre le stress oxydatif sont dépendants d’un apport adéquat en cuivre [
1]. Le Tableau I recense les principales cuproenzymes localisées dans les cellules humaines, leurs fonctions biologiques, ainsi que la nature des troubles associés à une perturbation de leur activité comme, par exemple, les maladies génétiques de Menkes et de Wilson [
2]. Par ailleurs, une augmentation inappropriée de la concentration en cuivre cellulaire peut mener à la production de radicaux hydroxyles via la réaction suivante :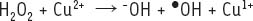
 | Tableau I.
Rôles de protéines cuivre-dépendantes dans la physiologie cellulaire.
|
Les radicaux hydroxyles réagissent avec les lipides, les protéines et l’ADN, engendrant ainsi des modifications qui altèrent leurs fonctions. Une concentration cellulaire adéquate en cuivre doit donc être maintenue afin d’éviter aussi bien une déficience nutritionnelle que la toxicité que risquerait de provoquer leur surcharge. Les organismes ont développé divers mécanismes pour incorporer le cuivre, le distribuer ou le séquestrer en cas d’excès. Nous discuterons de différents mécanismes d’acquisition et de distribution du cuivre d’après les connaissances acquises à partir d’études entreprises avec les levures Saccharomyces cerevisiae et Schizosaccharomyces pombe. |
Le transport de haute affinité du cuivre La plupart du temps, le cuivre circule sous sa forme oxydée (Cu2+). C’est sous sa forme réduite par des métalloréductases dans la membrane plasmique qu’il est pris en charge par des protéines de la famille des transporteurs de cuivre nommées Ctrs
(Figure 1).
 | Figure 1.
Membres de la famille des transporteurs Ctrs. Les transporteurs de la famille Ctr possèdent trois domaines transmembranaires, nommés TM1, TM2 et TM3 ( orange). La région amino-terminale extracellulaire des Ctr s est généralement riche en résidus méthionines. Ces AA forment les motifs Met ( MxxM et/ou MxM) ( rouge) qui sont reconnus pour leur potentiel à capter le cuivre extracellulaire. Tous les Ctr s illustrés ci-contre contiennent des motifs Met, sauf le transporteur humain/murin Ctr2 et celui nommé Ctr3 chez S. cerevisiae. Ctr3 est une protéine riche en résidus cystéines. Sur les onze cystéines que comporte le Ctr3, la mutation de quatre d’entre elles ( C) ( vert) altère sa fonction de transport du cuivre [
27]. La majorité des transporteurs Ctr s répertoriés jusqu’à présent, compte un résidu méthionine ( M) ( bleu) situé à environ vingt AA en amont du TM1. Ce résidu méthionine pourrait jouer un rôle important dans la coordination du cuivre près de la membrane plasmique, soit l’étape précédant le passage du cuivre à travers la membrane. Les transporteurs Ctr s présentent également une séquence MxxxM ( noir) à l’intérieur du TM2. Il semble que le cuivre soit coordonné par ces AA durant le processus de transport à travers la membrane. Il existe un motif composé de deux résidus glycines espacés de trois résidus ( GxxxG) ( mauve) à la hauteur du TM3. Ce motif, nommé GG4, est reconnu pour stabiliser l’interaction hélice-hélice à l’intérieur de la bicouche lipidique composant les membranes et permettre la multimérisation des protéines membranaires [
28]. |
Chez S. cerevisiae, les protéines Ctr1 et Ctr3 ont une forte affinité pour le cuivre (KM ~ 1-5 µm) et assurent son entrée dans la cellule. Les protéines Ctr1 et Ctr3 forment des homotrimères [
3]. Leurs actions sont indépendantes et redondantes. Un autre transporteur, Ctr2, est localisé à la surface de la vacuole [
4]. Devant une demande accrue pour le cuivre, ce transporteur fait passer l’ion de l’organite vers le cytosol afin de permettre l’approvisionnement en cuivre des protéines cytosoliques. Chez S. pombe, la prise en charge du cuivre par les transporteurs de haute affinité dépend également de la réduction du Cu2+ en Cu1+. La présence de protéines de la famille des Ctrs, Ctr4 et Ctr5 est requise pour le transport de l’ion [
5]. Contrairement aux autres Ctrs répertoriées, Ctr4 et Ctr5 forment un hétérocomplexe dans le sentier de sécrétion ; cette étape est primordiale pour sa localisation par la suite dans la membrane plasmique [5,
6]. Les régions de Ctr4 et Ctr5 qui font face au milieu extracellulaire présentent respectivement 5 et 2 motifs composés de résidus méthionines (appelés motifs Met). La présence de motifs Met, dans l’une ou l’autre des protéines, est suffisante pour l’incorporation du cuivre chez S. pombe [6]. Un troisième transporteur nommé Ctr6 est présent dans la membrane vacuolaire [
7]. Son action est similaire à celle de la protéine Ctr2 chez S. cerevisiae. Chez l’homme, le cuivre absorbé par les entérocytes est distribué, via la circulation sanguine, dans tout l’organisme. Les entérocytes, tout comme la plupart des cellules humaines, expriment le gène hCTR1, dont le produit est un transporteur de la famille Ctr [
8]. Ce transporteur (hCtr1) a d’ailleurs pu être repéré grâce à son homologie de séquence avec le transporteur Ctr1 de la levure. D’ailleurs, l’expression de hCTR1 a pour effet de rétablir l’entrée de cuivre dans une souche de levure déficiente en transporteurs Ctr1 et Ctr3. Chez l’homme, un second transporteur de la famille Ctr, nommé hCtr2, a été détecté [8]. Toutefois, sa fonction cellulaire demeure incertaine. Les études chez la souris prouvent à quel point l’incorporation du cuivre est capitale pour le développement de l’organisme. Les souris qui ont subi l’inactivation des deux allèles CTR1 codant pour le transporteur responsable de l’assimilation du cuivre meurent avant d’atteindre le milieu de la phase embryonnaire [
9]. Comme pour le transporteur humain, le Ctr1 murin a tout d’abord été caractérisé à la suite de son expression chez la levure. |
La distribution intracellulaire des ions cuivre Chez S. cerevisiae, il a été démontré que chaque cellule compte moins d’un ion de cuivre libre [
10]. En raison de cette rareté, le cuivre intracellulaire est associé à différentes protéines qui le distribuent et l’utilisent (Figure 2). Trois protéines chaperonnes spécifiques pour chacune de leur cible permettent la distribution du cuivre : Ccs1 [10], Cox17 [
11] et Atx1 [
12].
 | Figure 2.
Modèle de la distribution cellulaire du cuivre chez S. cerevisiae. La forme oxydée du cuivre (Cu2+
) est réduite par les réductases Fre1 et Fre2. Cette première étape est suivie du transport de l’ion par les protéines Ctr1 et Ctr3. Une fois à l’intérieur de la cellule, Ccs1 assure l’apport en cuivre à la SOD1. Cox17 est un élément-clé pour l’incorporation de cuivre à la CcO mitochondriale via les protéines Sco1 et Cox11. Atx1 permet l’approvisionnement en cuivre à l’appareil de Golgi de concert avec Ccc2. Dans le sentier de sécrétion, le cuivre est incorporé à diverses métalloprotéines telle la multi-cuivre oxydase Fet3. Dans ce cas précis, une fois le cuivre incorporé à Fet3, cette dernière est sécrétée à la surface cellulaire d’où elle peut catalyser le transport du Fe à l’intérieur de la cellule. Son action s’effectue en compagnie des réductases Fre1 et Fre2, ainsi que de la perméase Ftr1, à laquelle Fet3 est physiquement associée. Ctr2 est un transporteur vacuolaire de cuivre permettant de larguer l’ion dans le cytosol. Les astérisques indiquent les protéines de la levure pour lesquelles des protéines orthologues humaines ont été identifiées. Les noms de ces dernières sont indiqués entre parenthèses. RE : réticulum endoplasmique ; IMS : espace intermembranaire mitochondrial. |
L’apport en cuivre à la superoxyde dismutase 1 (SOD1) par la chaperonne Ccs1 La SOD1 fait partie de la majorité des organismes. Elle représente un élément-clé du processus de défense contre le stress oxydatif. Cet enzyme de faible masse moléculaire forme des homodimères. Chaque monomère contient un ion de zinc qui joue un rôle structural et un ion de cuivre qui agit en tant que cofacteur catalytique. La SOD1 catalyse la désagrégation des anions superoxydes en peroxyde d’hydrogène et en oxygène selon les réactions suivantes : 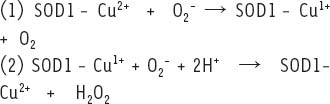
Chez S. cerevisiae, sans la chaperonne Ccs1, il n’y a pas d’incorporation de cuivre à l’enzyme SOD1 qui est toujours synthétisée mais demeure inactive. La chaperonne Ccs1 a été divisée en trois domaines selon des fragments polypeptidiques obtenus après une digestion partielle à la trypsine [
13]. Le premier domaine contient un motif consensus de liaison au Cu1+, MxCxxC, également présent chez la chaperonne Atx1. Le domaine I de Ccs1 peut agir en trans avec les domaines II et III pour favoriser l’apport en cuivre à la SOD1. Même si Atx1 montre une ressemblance avec le domaine I de Ccs1, cette chaperonne ne peut pas jouer un rôle équivalent en ce qui a trait à l’apport en cuivre à la SOD1. Le second domaine de Ccs1 partage une homologie de séquence avec la SOD1 et permet une interaction directe avec cet enzyme. La formation de l’hétérodimère Ccs1/SOD1 permet le transfert de l’ion Cu1+ d’une protéine à l’autre. Deux acides aminés (AA) essentiels à la dimérisation de la SOD1 figurent aussi dans le domaine II de Ccs1. La mutation de ces AA chez Ccs1 empêche l’interaction Ccs1/SOD1 et rend l’activité SOD1 nulle [13]. Le troisième domaine de la chaperonne Ccs1 est très conservé d’une espèce à l’autre et il est indispensable à l’action de Ccs1. Il contient un motif CxC essentiel à la liaison du Cu1+. Le cytosol constitue un environnement réducteur où très peu de protéines y forment des ponts disulfures. C’est pourtant le cas de la SOD1. Ce pont disulfure joue un rôle important dans le maintien d’une structure optimale pour son activité. Chez S. cerevisiae, la chaperonne Ccs1 permet non seulement l’apport en cuivre à la SOD1 mais reconnaît la protéine SOD1 immature, sans pont disulfure et sans cuivre. Par la suite, un pont disulfure transitoire relie la Cys-57 de la SOD1 et l’une des cystéines du motif CxC de Ccs1. Le pont disulfure intramoléculaire observé chez la SOD1 serait formé à la faveur de cet hétérocomplexe [
14]. Bien que les protéines SOD1 et Ccs1 soient pour la plupart localisées dans le cytosol, environ 1-5 % des protéines SOD1 sont localisées dans l’espace intermembranaire de la mitochondrie (IMS) [
15]. Dans l’IMS, la SOD1 est importée seulement sous sa forme immature, sans zinc, ni cuivre, ni pont disulfure et son importation ne dépend pas de la présence de Ccs1. Cependant, la séquestration de la SOD1 mature dans l’IMS est influencée par la localisation de Ccs1 dans ce compartiment cellulaire [
16]. La présence d’un pool de cuivre, non lié à des protéines, a récemment été mise en évidence dans la matrice mitochondriale. Cette source de cuivre pourrait servir à nourrir les enzymes dépendants du cuivre de la mitochondrie [
17]. Chez S. pombe, l’orthologue de la chaperonne Ccs1 de S. cerevisiae (ScCcs1) a été nommé Pccs [
18]. Chez les mammifères incluant l’homme, elle se nomme Ccs1. La chaperonne humaine acquiert le cuivre en liant ce dernier via ses domaines I et III. L’holo-molécule adopterait alors une conformation protégeant l’ion de cuivre jusqu’à son transfert à la SOD1. La passation du Cu de Ccs1 au site actif de la SOD1 relèverait du domaine III ; son domaine II quant à lui servirait de point d’arrimage avec la SOD1 [
19]. Une particularité de la Ccs1 humaine est qu’elle peut être convertie par une simple mutation ponctuelle en une protéine ayant une activité superoxyde dismutase comparable à celle de la SOD1. Les souris qui ont subi l’inactivation des deux allèles codant pour Ccs1 montrent une diminution significative de l’incorporation de Cu à la SOD1 et ce, particulièrement dans les cellules neuronales [
20]. L’hypothèse selon laquelle la chaperonne Ccs1 serait une des causes de certaines formes de sclérose latérale amyotrophique est aujourd’hui encore considérée. L’activation de la cytochrome c oxydase dans la mitochondrie L’apport en cuivre à la CcO de la membrane interne de la mitochondrie (IM) est la voie de distribution du cuivre la plus complexe (Figure 3). La CcO est cruciale pour la production d’énergie via la phosphorylation oxydative. Cette enzyme est composée de plus de douze sous-unités protéiques à l’intérieur desquelles agissent divers cofacteurs tels le cuivre, l’hème et le magnésium. Les trois plus grosses sous-unités de CcO sont Cox1, Cox2 et Cox3. La CcO requiert trois ions de cuivre pour être active : deux au centre Cu A, situé dans la partie de la protéine qui émerge de l’IMS pour Cox2 ; un au centre Cu B qui se trouve dans l’IM de la mitochondrie pour Cox1 [
21].  | Figure 3.
Modèle d’approvisionnement en cuivre pour l’activation de la CcO chez S. cerevisiae. Des travaux ont montré la présence d’un pool de cuivre à l’intérieur de la matrice mitochondriale [ 17]. Le cuivre présent dans la matrice correspond à ~85 % du cuivre mitochondrial. Dans la matrice, le cuivre serait lié à un ligand inconnu non-protéique de faible masse moléculaire [
29]. Le pool de cuivre pourrait agir comme source pour les cuproprotéines de l’IMS. Cependant, les molécules enrôlées dans le transport du cuivre vers l’IMS à partir de la machinerie de transport de haute affinité (Fre1, Fre2, Ctr1 et Ctr3) ou participant à l’acquisition de cet ion au niveau de la matrice mitochondriale, ne sont pas encore identifiées. C’est sous la forme « apo » que Cox17 est importée dans l’IMS via le complexe Tom. Une fois dans l’IMS, Cox17 pourrait prendre une forme tétramérique ou monomérique. La forme tétramérique servirait à séquestrer des ions de cuivre. La forme monomérique serait la forme lui permettant de concéder son cuivre à Sco1 et Cox11. Ces deux dernières protéines peuvent à leur tour fournir le cuivre aux sous-unités Cox1 et Cox2 de la CcO. Les astérisques indiquent les protéines de la levure pour lesquelles des protéines orthologues humaines ont été identifiées. OM : membrane externe ; IM : membrane interne ; IMS : espace intermembranaire mitochondrial. |
Le système d’approvisionnement en cuivre à la CcO n’est pas encore totalement défini. Le cuivre doit parvenir à la mitochondrie où les protéines Cox1 et Cox2 sont synthétisées. Chez S. cerevisiae, trois protéines contribuent à l’apport en cuivre à la CcO : Cox17, Cox11 et Sco1 [21]. Cox17 est une protéine soluble très conservée à travers les espèces eucaryotes qui vivent en aérobie, incluant l’homme. Cox17 possède la capacité de se lier au cuivre de façon réversible. Chez S. cerevisiae, une souche mutante pour COX17 présente une CcO inactive. Le modèle initial suggérait que Cox17 contribuait à l’apport en cuivre à l’intérieur de la mitochondrie [11] bien que l’absence de Cox17 n’affecte pas le niveau de cuivre contenu dans la mitochondrie [21]. De plus, l’environnement réducteur du cytosol laisserait Cox17 sous sa forme réduite, permettant plus facilement son entrée dans l’IMS via le complexe Tom [21]. Des études récentes suggèrent que la forme active de Cox17 opère dans l’IMS. C’est à cet endroit que la protéine acquerrait le cuivre. Cette étape serait alors suivie de l’apport de l’ion métallique par Cox17 aux protéines Sco1 et Cox11 [
22]. La protéine Sco1, requise pour l’incorporation du cuivre au site CuA de la sous-unité Cox2, est ancrée à l’IM de la mitochondrie via un domaine transmembranaire. Elle interagit directement avec Cox2 [21]. Sco1 et Cox2 partagent une homologie de séquence des motifs (CxxxC) en action dans la coordination du Cu1+. Lorsque le gène SCO1 est inactivé, la sous-unité Cox2 est rapidement dégradée. Il existe une protéine homologue à Sco1, nommée Sco2 [21]. Chez S. cerevisiae, cette protéine n’est pas essentielle à l’apport en cuivre à la CcO. En revanche, lorsqu’elle est surexprimée, Sco2 peut partiellement substituer la fonction de la protéine Sco1 dans l’apport en cuivre à la CcO [21]. Chez l’homme, il a été démontré que Sco1 et Sco2 sont toutes deux requises pour l’incorporation du cuivre au site CuA de Cox2 [
23]. De plus, bien que les deux protéines Sco1 et Sco2 soient exprimées de façon ubiquitaire dans tous les tissus et dans tous les types cellulaires examinés, leurs actions sont indépendantes et non-redondantes [23]. De fait, les mutations affectant le gène SCO1 ne provoquent pas les mêmes symptômes cliniques que ceux causés par des mutations touchant le gène SCO2. Les rôles respectifs à l’échelle moléculaire de Sco1 et Sco2 ne sont toujours pas élucidés. La protéine Cox11 est une composante ancrée à l’IM via un domaine transmembranaire. Cox11 forme un dimère et lie un ion de Cu1+ par monomère. La protéine Cox11 est impliquée dans l’apport en cuivre au site CuB de la sous-unité Cox1. D’ailleurs, Cox11 interagit directement avec cette dernière sous-unité [22]. L’approvisionnement en cuivre à l’appareil de Golgi Chez S. cerevisiae, la chaperonne Atx1 est localisée dans le cytosol. Atx1 contient un motif de liaison au cuivre de type MxCxxC [ 12]. Cette chaperonne assure l’apport en cuivre à un transporteur intracellulaire situé dans la membrane de l’appareil de Golgi nommé Ccc2 [
24]. Huit domaines transmembranaires ancrent Ccc2 à la membrane de l’appareil de Golgi. La région cytosolique amino-terminale de Ccc2 présente également deux motifs de liaison au cuivre de type MxCxxC. Des travaux ont démontré qu’Atx1 interagit directement avec la section amino-terminale de Ccc2. Cette interaction est dépendante de la présence de cuivre, prévenant ainsi les interactions improductives entre la forme « apo » de Ccc2 et Atx1 [
25]. La protéine Atx1 et la région amino-terminale de Ccc2 possèdent des structures tertiaires similaires bien qu’existe une exposition d’AA de charges négatives chez Ccc2 et d’AA de charges positives chez Atx1 à l’interface de leurs zones d’interactions respectives. La mutation de ces AA chez Atx1 abolit l’interaction Atx1/Ccc2. Le transfert du cuivre dans l’appareil de Golgi requiert l’hydrolyse d’ATP par Ccc2. Une fois dans l’appareil de Golgi, le cuivre est incorporé à des protéines destinées à être sécrétées ou acheminées vers différents compartiments cellulaires. Chez S. cerevisiae, l’incorporation du cuivre à la protéine Fet3 a lieu au sein de l’appareil de Golgi. Par la suite, Fet3 est acheminée à la membrane plasmique (Fet3 est l’une des protéines qui composent le système d’acquisition du fer de haute affinité). D’ailleurs, chez des souches mutantes pour les gènes ATX1 et CCC2, le transport de haute-affinité du fer est aboli. Chez l’homme, il existe un homologue de la chaperonne Atx1 qui se nomme Atox1 [1], ainsi que deux homologues du transporteur Ccc2, soit ATP7A (protéine de Menkes) et ATP7B (protéine de Wilson) [2]. Des travaux ont montré que Atox1 interagit avec les transporteurs ATP7A et ATP7B. La protéine de Menkes est exprimée dans tous les types cellulaires, sauf dans les hépatocytes. Elle assure l’apport en cuivre à l’appareil de Golgi où certains enzymes acquièrent leur cofacteur : la dopamine β-hydroxylase, la peptidylglycine-mono-oxygénase, la superoxyde dismutase extracellulaire, et la tyrosinase (Tableau I). Dans les fibroblastes et les cellules épithéliales polarisées de l’intestin, la protéine de Menkes se déplace de l’appareil de Golgi vers la membrane plasmique lorsque la concentration en cuivre augmente. Le cuivre est alors expulsé de la cellule et passe dans la circulation sanguine. Si la concentration en cuivre baisse, la protéine de Menkes regagne l’appareil de Golgi. Elle joue donc un rôle très important dans la mobilisation du cuivre chez l’homme. Chez les patients atteints du syndrome de Menkes, les symptômes cliniques observés sont une dégénérescence cérébrale, un développement osseux inadéquat, des anomalies cardio-vasculaires accompagnées de difficultés à maintenir la température corporelle [1]. L’injection intraveineuse de cuivre couplé à l’histidine permet d’atténuer ces symptômes mais n’empêche pas toutefois la dégénérescence des cellules nerveuses [2]. Pour sa part, la protéine de Wilson est exprimée la plupart du temps dans l’appareil de Golgi des hépatocytes où elle permet l’incorporation du cuivre à la céruloplasmine (Tableau I). Lorsque la concentration en cuivre augmente dans la cellule, la protéine de Wilson se dirige vers des compartiments vésiculaires situés en périphérie de la membrane plasmique. Ces compartiments vésiculaires pourraient s’associer au canal biliaire pour excréter le cuivre excédentaire via la bile. La maladie de Wilson est caractérisée par l’accumulation d’une quantité excessive de cuivre dans le foie, la protéine ATP7B mutante ne pouvant évacuer l’excès de cuivre. Il s’ensuit une nécrose du foie qui provoque la libération du cuivre dans le système sanguin ; le cuivre s’accumule alors dans divers organes notamment le cerveau et les reins. Les patients atteints de cette maladie souffrent de cirrhose et de troubles neurologiques et psychiatriques [1]. |
Les études utilisant les organismes modèles unicellulaires comme la levure ont permis de découvrir des protéines de transport et de distribution des ions de cuivre [
26]. Les mécanismes en cause sont remarquablement bien orchestrés, permettant à la fois d’assurer les besoins en cuivre et d’éviter la toxicité qu’engendrerait une surcharge. Les études publiées ont établi la preuve que la levure constitue un excellent modèle pour comprendre les bases moléculaires des tares héréditaires auxquelles sont attribuées des défaillances métaboliques chez l’humain. Étant donné l’existence de composantes cuivre-dépendantes importantes en cause dans les maladies de Creutzfeldt-Jakob (protéine prion) et d’Alzheimer (protéine précurseur β-amyloïde), dans la sclérose latérale amyotrophique (SOD1) et dans l’acidose lactique (CcO), il devient primordial d’élucider les divers mécanismes et sentiers moléculaires empruntés par le cuivre afin d’atteindre ces métalloprotéines.

|
1. Madsen E, Gitlin JD. Copper and iron disorders of the brain. Annu Rev Neurosci 2007; 30 : 317–37. 2. La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys 2007; 463 : 149–67. 3. Puig S, Lee J, Lau M, Thiele DJ. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J Biol Chem 2002; 277 : 26021–30. 4. Rees EM, Lee J, Thiele DJ. Mobilization of intracellular copper stores by the Ctr2 vacuolar copper transporter. J Biol Chem 2004; 279 : 54221–29. 5. Zhou H, Thiele DJ. Identification of a novel high affinity copper transport complex in the fission yeast Schizosaccharomyces pombe. J Biol Chem 2001; 276 : 20529–35. 6. Beaudoin J, Laliberté J, Labbé S. Functional dissection of Ctr4 and Ctr5 amino-terminal regions reveals motifs with redundant roles in copper transport. Microbiology 2006; 152 : 209–22. 7. Bellemare DR, Shaner L, Morano KA, et al. Ctr6, a vacuolar membrane copper transporter in Schizosaccharomyces pombe. J Biol Chem 2002; 277 : 46676–86. 8. Zhou B, Gitschier J. hCTR1, a human gene for copper uptake identified by complementation in yeast. Proc Natl Acad Sci USA 1997; 94 : 7481–6. 9. Lee J, Prohaska JR, Thiele DJ. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc Natl Acad Sci USA 2001; 98 : 6842–47. 10. Rae TD, Schmidt PJ, Pufahl RA, et al. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 1999; 284 : 805–8. 11. Glerum DM, Shtanko A, Tzagoloff A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J Biol Chem 1996; 271 : 14504–9. 12. Lin SJ, Pufahl RA, Dancis A, et al. A role for the Saccharomyces cerevisiae ATX1 gene in copper trafficking and iron transport. J Biol Chem 1997; 272 : 9215–20. 13. Schmidt PJ, Rae TD, Pufahl RA, et al. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J Biol Chem 1999; 274 : 23719–25. 14. Furukawa Y, Torres AS, O’Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J 2004; 23 : 2872–81. 15. Sturtz LA, Diekert K, Jensen LT, et al. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, Ccs, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem 2001; 276 : 38084–9. 16. Field LS, Furukawa Y, O’Halloran, TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem 2003; 278 : 28052–9. 17. Cobine PA, Ojeda LD, Rigby KM, Winge DR. Yeast contains a non-proteinaceous pool of copper in the mitochondrial matrix. J Biol Chem 2004; 279 : 14447–55. 18. Laliberté J, Whitson LJ, Beaudoin J, et al. The Schizosaccharomyces pombe Pccs protein functions in both copper trafficking and metal detoxification pathways. J Biol Chem 2004; 279 : 28744–55. 19. Rae TD, Torres AS, Pufahl RA, O’Halloran TV. Mechanism of Cu, Zn-superoxide dismutase activation by the human metallochaperone hCCS. J Biol Chem 2001; 276 : 5166–76. 20. Subramaniam JR, Lyons WE, Liu J, et al. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci 2002; 5 : 301–7. 21. Cobine PA, Pierrel F, Winge DR. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim Biophys Acta 2006; 1763 : 759–72. 22. Horng YC, Cobine PA, Maxfield AB, et al. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome c oxidase. J Biol Chem 2004; 279 : 35334–40. 23. Leary SC, Cobine PA, Kaufman BA, et al. The human cytochrome c oxidase assembly factors Sco1 and Sco2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab 2007; 5 : 9–20. 24. Yuan DS, Stearman R, Dancis A, et al. The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc Natl Acad Sci USA 1995; 92 : 2632–6. 25. Banci L, Bertini I, Cantini F, et al. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat Chem Biol 2006; 2 : 367–8. 26. Rees EM, Thiele DJ. From aging to virulence: forging connections through the study of copper homeostasis in eukaryotic microorganisms. Curr Opin Microbiol 2004; 7 : 175–84. 27. Peña MMO, Puig S, Thiele DJ. Characterization of the Saccharomyces cerevisiae high affinity copper transporter Ctr3. J Biol Chem 2000; 275 : 33244–51. 28. Aller SG, Eng ET, De Feo CJ, Unger VM. Eukaryotic Ctr copper uptake transporters require two faces of the third transmembrane domain for helix packing, oligomerization, and function. J Biol Chem 2004; 279 : 53435–41. 29. Cobine PA, Pierrel F, Bestwick ML, Winge DR. Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J Biol Chem 2006; 281 : 36552–9. |





