| |
| Med Sci (Paris). 2008 June; 24(6-7): 629–634. Published online 2008 June 15. doi: 10.1051/medsci/20082467629.Inférence des réseaux de régulation transcriptionnelle Xavier Gidrol,* Ning Wu, Vincent Frouin, and Marie-Anne Debily CEA, DSV, IRCM, Laboratoire d’Exploration Fonctionnelle des Génomes, 2, rue Gaston Crémieux, CP22, 91057 Évry Cedex, France |
La cellule vivante est un système extrêmement complexe. Des dizaines de milliers de gènes, des centaines de milliers de transcrits, des millions de protéines, des dizaines de millions de petites molécules interagissent entre elles, à chaque instant au sein de la cellule pour réguler ses fonctions. Que ce soit sous le vocable de génomique fonctionnelle, de biologie intégrative ou de biologie systémique, la compréhension des réseaux d’interactions moléculaires est devenue l’un des grands enjeux de la biologie cellulaire en ce début de siècle. Il existe plusieurs types de réseaux biologiques, qui, comme pour les autres types de réseaux - réseau Internet, réseaux ferroviaires, réseaux sociaux, etc. - sont représentés essentiellement par des nœuds, qui sont les objets biologiques étudiés, et des liens ou arcs qui définissent les interactions entre les nœuds du réseau pour une cellule dans un état donné. Les réseaux métaboliques regroupent toutes les réactions enzymatiques et répertorient pour chaque enzyme, les substrats et les produits de la réaction. Dans ces réseaux, les nœuds sont les métabolites et les étiquettes des arcs sont les enzymes. Les réseaux métaboliques sont très bien définis en terme topologique, mais aussi en terme quantitatif, grâce à une connaissance fine des propriétés des enzymes et de leurs paramètres cinétiques (Km, Vm, constantes d’association ou d’inhibition, etc.) acquis par des milliers de biochimistes et d’enzymologistes dans le monde depuis plusieurs décennies. La description des réseaux d’interaction « protéine-protéine » est fondée essentiellement sur la technologie du double hybride, même si depuis peu, d’autres approches utilisant la complémentation de fragments protéiques et la reconstitution d’activités enzymatiques [
1] permettent aussi d’étudier ce type d’interactions. À l’inverse des réseaux métaboliques, les nœuds du réseau sont les protéines et les arcs symbolisent l’existence d’une interaction entre deux protéines, sans plus d’information sur sa nature, ses règles ou son rôle dans la cellule. Ces réseaux sont donc très peu informatifs sur la fonctionnalité des interactions. Les réseaux de régulation transcriptionnelle ou réseaux génétiques, ont un niveau de complexité plus élevé encore. Ils constituent de facto des réseaux de réseaux, car ils intègrent certaines voies métaboliques, comme les cascades de kinases par exemple, des interactions protéine-protéine, par exemple l’interaction de la protéine E2F avec la protéine RB (Rétinoblastome), et des interactions plus spécifiques, entre protéines et ADN, comme par exemple la fixation d’un facteur de transcription sur sa séquence régulatrice. Dans les réseaux de régulation transcriptionnelle, les nœuds représentent indifféremment des gènes ou des protéines, les arcs définissent le rôle d’un gène sur l’expression d’un second. Une règle, telle que l’induction ou la répression du gène y par le gène x, peut être associée à un arc. Dans cet article, nous évoquerons uniquement les réseaux de régulation transcriptionnelle pour deux raisons essentiellement. La première est qu’il s’agit d’un domaine où de grandes quantités de données peuvent être générées, grâce aux approches à haut débit de la génomique fonctionnelle comme les puces à ADN. La deuxième est que l’expression des gènes est souvent l’origine et l’effecteur d’une réponse cellulaire. C’est un point dans l’espace et le temps cellulaires où l’information contenue dans le génome est décodée et interprétée pour fabriquer les protéines requises pour une action cellulaire. Nous décrirons les développements récents en génomique fonctionnelle, qui permettent, grâce à l’intégration des données générées, d’inférer les réseaux de régulation transcriptionnelle. |
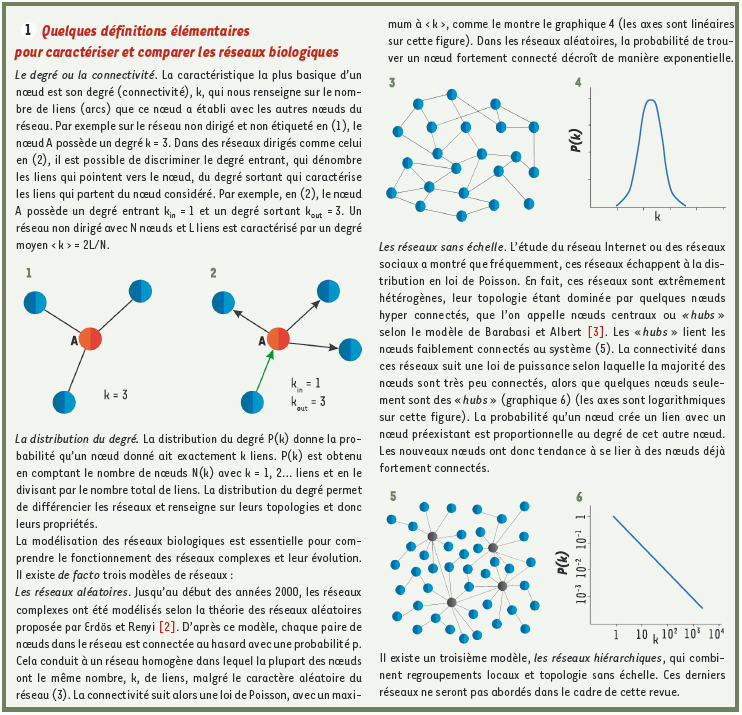
Propriétés des réseaux biologiques Comprendre la topologie et la dynamique des réseaux biologiques devrait nous permettre à terme de définir leurs propriétés : leur rôle dans les réponses cellulaires engendrées par des perturbations intrinsèques ou extrinsèques, leur stabilité par rapport à ces perturbations et enfin leur impact sur l’établissement d’un phénotype cellulaire. La plupart des réseaux biologiques partagent de nombreuses propriétés avec les autres types de réseaux. Les réseaux biologiques, comme les réseaux sociaux ou Internet, sont sans échelle, c’est-à-dire que le degré de connectivité d’un nœud (le nombre d’arcs connectés à ce nœud) suit une distribution en loi de puissance (Encadré 1). Il s’ensuit donc que la majorité des nœuds sont très peu connectés, alors que quelques nœuds sont au contraire hyper-connectés et agissent comme des nœuds centraux ou hubs. Ces nœuds centraux joueraient un rôle prépondérant pour conduire la réponse de la cellule à une perturbation. La rareté des nœuds centraux donne donc de la stabilité et de la robustesse au réseau. Par ailleurs, la plupart des réseaux biologiques sont qualifiés de « petit monde », c’est-à-dire qu’il faut très peu d’arcs pour connecter n’importe quels nœuds du réseau entre eux. Le très grand nombre de relations possibles entre nœuds, via leurs voisins, est à rapprocher de la situation observée dans les réseaux sociaux où les individus se découvrent des relations communes (« le monde est petit !… »). Enfin, alors que pour les réseaux sociaux l’on observe des connexions accrues entre les nœuds centraux, il semble que ce ne soit pas le cas pour les réseaux biologiques. Cette originalité pourrait s’expliquer par la nécessité d’une compartimentation entre certaines grandes fonctions cellulaires parfois opposées, prolifération et différenciation par exemple. Ou bien encore parce qu’il existerait une limite de connexions possibles sur un nœud central, sous peine de fragilisation excessive du réseau. L’évolution n’aurait donc pas sélectionné ce type de nœuds centraux très connectés entre eux. Les lecteurs souhaitant se documenter de manière plus exhaustive sur les propriétés des réseaux biologiques pourront consulter l’excellente revue de Barabasi et Oltvai [
4]. |
Inférence des réseaux de régulation transcriptionnelle La complexité des réseaux de régulation transcriptionnelle chez les métazoaires reflète la multitude des fonctions qu’ils régulent et rend leur caractérisation extrêmement difficile. Pour comprendre la topologie d’un réseau et sa dynamique, il est nécessaire d’intégrer diverses approches de biologie à grande échelle [
18]. Ainsi pour caractériser un réseau il convient de : (1) définir l’identité des nœuds du réseau et caractériser leur niveau d’expression ; (2) étudier la dynamique des interactions en fonction du temps comme par exemple au cours du cycle cellulaire ou de la différenciation ; (3) étudier le comportement du réseau, notamment sa robustesse, en réponse à des perturbations. À ce titre, l’extinction systématique des nœuds d’un réseau par ARN interférence et l’analyse de son impact sur le phénotype cellulaire est extrêmement utile. L’intérêt de ces approches à haut débit est leur complémentarité. Il est en effet possible de générer des informations complémentaires, qui éventuellement se recoupent, pour pallier les faiblesses de chacune des approches prise isolément. Ainsi, dans notre équipe nous utilisons une démarche expérimentale, schématisée sur la Figure 1, fondée sur l’intégration de diverses approches à grande échelle ; l’analyse du transcriptome par puce à ADN, l’immunoprécipitation de chromatine sur puce (ChIP on chip) et les cribles fonctionnels par ARN interférence à haut débit.  | Figure 1.
Une approche systématique et intégrative pour inférer le réseau de régulation transcriptionnelle d’un facteur de transcription (FT) particulier. Afin d’identifier les gènes régulés par FT, des cellules délétées (cellules FT−/−) ou non (cellules sauvages) pour ce FT peuvent être utilisées pour analyser les modulations résultantes au niveau transcriptionnel (données transcriptome) et au niveau des cibles génomiques (données ChIP on chip). L’intégration de ces données génomiques peut alors permettre de construire la topologie du réseau en identifiant les gènes régulés par ce FT et d’étudier la dynamique de ce réseau, notamment via l’utilisation de séries temporelles. Pour identifier les régulateurs du FT, une stratégie différente est envisagée. Des cellules contenant une construction reportrice dans laquelle le promoteur du FT est fusionné à un gène rapporteur sont utilisées pour cribler à haut débit des collections de siARN sur puce. L’identification des gènes dont l’extinction entraîne une induction ou une répression de l’expression du FT permet de compléter la topologie du réseau, en identifiant les gènes régulant le FT. L’ensemble des informations collectées sur la topologie du réseau peut également servir de base pour identifier les partenaires clés du FT dans le réseau. Ces partenaires peuvent à leur tour être éliminés et servir à générer de nouvelles données génomiques qui viendront enrichir la topologie du réseau établie précédemment. L’impact du réseau sur le phénotype des cellules est étudié par extinction systématique des nœuds du réseau. |
|
Immunoprécipitation de chromatine sur puce ( ChIP on chip) La technologie des puces à ADN, abondamment décrite dans la littérature [
5–
8], permet d’obtenir des profils d’expression, c’est-à-dire de mesurer la concentration à l’équilibre de la totalité des ARNm d’une cellule. Toutefois, un changement de concentration à l’équilibre est parfois abusivement assimilé à un accroissement de la vitesse de transcription ou même à une modification du niveau de protéines traduites à partir de cet ARNm, alors que ce changement de concentration pourrait résulter simplement d’un ralentissement de la dégradation de l’ARNm, donc de l’augmentation de sa demi-vie, sans que sa transcription ait été affectée. Pour compléter les données d’expression, préciser si la transcription est affectée et extraire des informations quantitatives supplémentaires, la technologie de « ChIP on chip » récemment proposée [
9] offre des perspectives intéressantes. Développée initialement chez la levure, elle permet d’étudier in vivo tous les sites de fixation d’un facteur de transcription sur un génome et complète ainsi les données du « transcriptome » [
10,
11]. Elle présente donc de nombreux avantages pour l’analyse des réseaux génétiques. En effet, les études de ChIP on chip se font à partir de cellules tout à fait normales, caractérisées par une expression constitutive et naturelle des facteurs de transcription. Ensuite, la méthode mesure uniquement les interactions directes entre les protéines régulatrices et leurs cibles. Ce point est particulièrement important car il permet d’étudier le nombre d’intermédiaires entre un facteur de transcription et ses cibles, contrairement aux profils d’expression ou aux expériences basées sur la génétique qui ne permettent pas de distinguer les effets directs des effets indirects. Enfin, la technologie de ChIP on chip permet d’identifier des sites de fixation des facteurs de transcription dont les séquences nucléotidiques sont très proches, ou, à l’inverse, de caractériser des sites tellement différents qu’il n’existe pas de vraie séquence consensus entre eux. Or ce n’est pas le cas des approches purement algorithmiques in silico de détermination de séquences régulatrices, qui ont parfois du mal à distinguer des sites de fixation de facteurs de transcription d’une même famille ayant des séquences très proches.
|
L’utilisation de l’ARN interférence systématique Pour compléter l’étude d’un réseau, l’ARN interférence à haut débit appliquée à l’ablation systématique de chaque nœud d’un réseau est une méthode de choix. Elle permet en effet de tester la robustesse d’un réseau et définir ainsi les nœuds qui stabilisent, ou au contraire, sensibilisent le réseau. L’ARN interférence a beaucoup d’avantages par rapport aux méthodes conventionnelles utilisées pour supprimer l’expression d’un gène [
12,
13,
19]. Elle est adaptable au crible à haut débit et l’ablation transitoire « non chronique » d’un facteur de transcription peut permettre d’éviter les phénomènes compensatoires, les redondances fonctionnelles, que l’on observe parfois chez les souris knock-out. Les puces à siARN que nous développons dans notre laboratoire [
14,
15] permettent par exemple d’étudier tous les gènes qui régulent l’activité d’un promoteur grâce à une extinction systématique de tous les gènes humains pouvant contrôler la régulation de l’expression de ce gène d’intérêt. Le principe de l’étude est d’utiliser des cellules ayant intégré une construction contenant le promoteur du gène X fusionné à un gène rapporteur, afin de cribler les gènes activateurs ou répresseurs de la transcription du gène X (Figure 2A). Pour réaliser ce crible à haut débit, nous avons développé une puce à siARN permettant de transfecter des cellules simultanément avec des milliers de siARN et d’analyser l’influence de chaque siARN sur cette construction utilisée comme « rapporteur » (Figure 2B).  | Figure 2.
Identification des régulateurs transcriptionnels d’un facteur de transcription particulier par puce à siARN. A. Principe du crible. Des cellules contenant une construction reportrice dans laquelle le promoteur du gène FT est fusionné à un gène rapporteur, en l’occurrence l’eGFP (enhanced green fluorescence protein), sont produites. Les cellules présenteront donc un niveau d’expression de l’eGFP qui reflète le niveau basal d’expression du gène FT dans un type de cellules donné. Le niveau d’expression de l’eGFP peut être facilement mesuré à l’aide d’un scanner permettant de détecter l’émission de fluorescence résultant de l’excitation de la GFP. La transfection parallèle dans ces cellules de quantités massives de siARN permet d’identifier les régulateurs du gène FT. En effet, les gènes cibles des siARN entraînant une augmentation du niveau d’expression de la GFP, correspondront à des régulateurs négatifs du gène FT. À l’inverse une diminution de l’expression de la GFP indiquera que le gène ciblé par le siARN agit comme un régulateur positif du gène FT dans la cellule. B. Principe des puces. Les collections de siARN sont déposées de manière ordonnée sur une lame de verre à l’aide d’un robot. Chaque dépôt correspond à un mélange d’un siARN particulier complexé à un agent de transfection et à un polymère. Les cellules peuvent alors être cultivées sur la lame de verre de telle sorte qu’au niveau de chaque dépôt les cellules seront transfectées par un siARN particulier. Après 2 à 3 jours de culture, le phénotype résultant de l’ARN interférence pourra être analysé en terme d’augmentation (spot rouge), de diminution (spot blanc) ou d’absence de modulation (spot vert) du niveau d’expression de l’eGFP. |
L’extinction de gènes nous sert également à comparer les profils d’expression et les cibles identifiés par ChIP on chip dans des cellules sauvages pour le facteur de transcription considéré et dans des cellules dans lesquelles l’expression de ce facteur de transcription a été drastiquement réduite. À partir de ces mêmes cellules nous analysons les phénotypes pertinents par rapport à la question biologique initialement posée, prolifération ou différenciation par exemple. À partir de la connaissance de la topologie du réseau et de sa dynamique, des prédictions sur le phénotype peuvent même être faites et vérifiées expérimentalement. |
Même si les réseaux de régulation transcriptionnelle restent excessivement complexes à étudier, des technologies et des stratégies expérimentales se mettent en place, pas à pas, pour inférer leur topologie et étudier leur dynamique. L’objectif à très long terme serait de pouvoir prédire « l’attracteur » d’un réseau, c’est-à-dire son prochain état stable en réponse à une perturbation (Encadré 2). Cela pourrait être, par exemple, la caractérisation des réseaux de régulation transcriptionnelle mis en place au cours de l’initiation et de la progression tumorales. Comprendre ces réseaux permettrait peut-être de pouvoir un jour les rediriger vers « l’attracteur » de notre choix et de limiter l’initiation ou la progression tumorales.

|
1. Michnick SW, Ear PH, Emily, et al. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov 2007; 6 : 569–82. 2. Erdos P, Renyi A. On the evolution of random graphs. Math Inst Hungarian Acad Sci 1960; 5 : 17–61. 3. Barabasi AL, Albert R. Meergence of scaling in random network. Science 1999; 286 : 509–12. 4. Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet 2004; 5 : 101–13. 5. Canales RD, Luo Y, Willey JC, et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotech 2006; 24 : 1115–22. 6. Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, et al. Performance comparison of one-color and two-color platforms within the microarray quality control (MAQC) project. Nat Biotech 2006; 24 : 1140–50. 7. Shi L, Reid LH, Jones WD, et al. The microarray quality control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotech 2006; 24 : 1151–61. 8. Sotiriou C, Piccart MJ. Taking gene-expression profiling to the clinic: when will molecular signatures become relevant to patient care ? Nat Rev Cancer 2007; 7 : 545–53. 9. Ren B, Robert F, Wyrick JJ, et al. Genome-wide location and function of DNA binding proteins. Science 2000; 290 : 2306–9. 10. Cawley S, Bekiranov S, Ng HH, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNA. Cell 2004; 116 : 499–509. 11. Vigano MA, Lamartine J, Testoni B, et al. New p63 targets in keratinocytes identified by a genome-wide approach. EMBO J 2006; 25 : 5105–16. 12. Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391 : 806–11. 13. Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001; 411 : 494–8. 14. Baghdoyan S, Roupioz Y, Pitaval A, et al. Quantitative analysis of highly parallel transfection in cell microarrays. Nucleic Acids Res 2004; 32 : e77. 15. Roupioz Y, Castel D, Pitaval A, et al. Puces à cellules et génomique fonctionnelle. Med Sci (Paris) 2005; 21 : 535–8. 16. Basso K, Margolin AA, Stolovitzky G, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet 2005; 4 : 382–90. 17. Balis A, Tsikitis M, Acosta-Alvear D, et al. An initial blueprint for myogenic differentiation. Genes Dev 2005; 19 : 553–69. 18. Martin D, Ghattas B, Thieffry D. Prédire la transcription à partir des séquences génomiques. Med Sci (Paris) 2004; 20 : 1036–40. 19. Dautry F, Ribet C. L’interférence par l’ARN : vers une génomique fonctionnelle chez les mammifères ? Med Sci (Paris) 2004; 20 : 815–9. |



