| |
| Med Sci (Paris). 2012 October; 28(10): 870–875. Published online 2012 October 12. doi: 10.1051/medsci/20122810016.Structure des récepteurs mu et delta des opiacés Sébastien Granier1* 1Institut de génomique fonctionnelle, Inserm, CNRS, UMR1 UMR2, 141, rue de la Cardonille, 34094Montpellier Cedex 5, France |
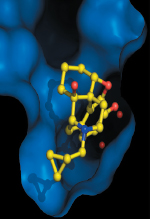
Photo : un antagoniste morphinique vu dans la poche de liaison du récepteur mu des opiacés (© Sébastien Granier). Les récepteurs des opiacés (OR) jouent un rôle clé dans le système nerveux central, régulant la perception de la douleur, la sensation de bien-être et de plaisir, et l’humeur [
1]. Ils appartiennent à la famille des récepteurs couplés aux protéines G (RCPG) et ont été classés en trois sous-types en fonction de leur pharmacologie et de leur distribution tissulaire : le mu-OR (avec pour ligand la morphine), le delta-OR (localisé au niveau des vas deferens) et le kappa-OR (avec pour agoniste le kétocyclazocine) [
2]. Ils sont impliqués dans l’action de la morphine, de l’héroïne, et de peptides endogènes comme les endorphines et les enképhalines, et sont donc des cibles pharmacologiques de choix pour le traitement des douleurs intenses. Pour mieux comprendre les bases moléculaires et structurales de la pharmacologie des OR, nous avons entrepris de déterminer leur structure tridimensionnelle par diffraction aux rayons X. Nous avons utilisé la technique de cristallisation in meso [
3] et déterminé les structures du mu-OR et du delta-OR liés aux antagonistes β-funaltrexamine et naltrindole, à une résolution de 2,8 et 3,4 Å, respectivement [
4,
5]. |
Vue d’ensemble de la structure des récepteurs aux opiacés mu et delta Les OR présentent une architecture à 7 domaines transmembranaires (7TM), une caractéristique commune à tous les RCPG (Figure 1A) [
13] (→).
(→) Voir la synthèse de G. Lebon et C.G. Tate, page 876 de ce numéro
 | Figure 1.
Vue d’ensemble des structures des récepteurs aux opiacés mu et delta. A. Les schémas du haut représentent une vue parallèle dans le plan de la membrane plasmique du mu-OR (en bleu) et du delta-OR (en orange), et montrent l’organisation structurale en 7 domaines transmembranaires (TM1 à TM7), avec 3 boucles extracellulaires (ECL1 à ECL3), et la présence de l’hélice 8. Les schémas du bas montrent une vue de dessus (côté extracellulaire) des récepteurs illustrant l’organisation des 7TM et des 3 ECL, ainsi que la position des ligands β-funaltrexamine (sphères vertes) et naltrindole (sphères jaunes). B,C. Comparaison de la structure de la boucle extracellulaire ECL1 (B) et ECL2 (C) de mu-OR et de delta-OR, ainsi que de leur séquence primaire. Les acides aminés qui diffèrent entre mu-OR et delta-OR sont en rouge. |
L’ossature structurale de ces domaines est très conservée entre mu-OR et delta-OR (Figure 1A). Les ligands antagonistes se positionnent dans une poche similaire (
Figure 1A
, schémas du bas), relativement enfouie dans les 7TM et largement ouverte vers le domaine extracellulaire. Cette accessibilité importante au site de liaison pourrait être à l’origine de l’extraordinaire rapidité d’action des opiacés. De même, elle pourrait expliquer la réversibilité des overdoses d’héroïne par le naloxone, un antagoniste utilisé en clinique. Les domaines transmembranaires des OR sont connectés par trois boucles au niveau extracellulaire (ECL1 à ECL3) [13]. Une observation intéressante concerne la conservation du repliement de ces boucles extracellulaires, et ceci en dépit d’une divergence de leur séquence primaire (Figure 1BC). Par exemple, la boucle ECL2, qui connecte le TM4 et le TM5, présente une structure secondaire en épingle à cheveux de type β dans tous les récepteurs aux opiacés [4–
7], comme illustré pour mu-OR et delta-OR dans la
Figure 1C
. |
La poche de liaison du récepteur mu-OR et delta-OR : un mode de liaison conservé Les ligands utilisés pour nos études sont des antagonistes de type morphinique. La
Figure 2A
présente l’organisation structurale des sites de liaison du naltrindole (en haut) et de la β-funaltrexamine (en bas). Le mode de liaison de ces composés est très similaire dans les deux structures avec des contacts hydrophobes, des interactions de type charge-charge et un réseau d’interaction de liaisons hydrogène (Figure 2). En particulier, l’Asp3.321 forme une interaction charge-charge avec le groupe amine des ligands morphiniques, et forme une liaison hydrogène avec la Tyr7.43 (Figure 2A). Les ligands opiacés peuvent être de nature différente (petites molécules ou peptides), mais beaucoup possèdent des caractéristiques communes, comme un groupe hydroxyphényl séparé d’une charge positive par six carbones mimant la partie amino-terminale de type tyrosine de tous les peptides opiacés endogènes (Figure 2B). Cette signature moléculaire est associée à un mode de liaison conservé dans tous les OR (Figure 2C) avec l’interaction charge-charge précédemment citée avec l’Asp 3.32, et des interactions hydrophobes avec la Tyr3.33, la Val6.55 et l’Ile6.51. De plus, dans les deux structures OR, nous avons observé la présence d’une densité électronique compatible avec deux molécules d’eau. Ces molécules d’eau sont bien positionnées pour former un réseau d’interaction de liaisons hydrogène connectant l’His6.52 avec le groupe hydroxyphényl du groupe morphinique (Figure 2C).
 | Figure 2.
La poche de liaison des récepteurs mu et delta des opiacés. A. Organisation structurale du site de liaison du naltrindole (schémas du haut) et de la β-funaltrexamine (schémas du bas). Le ligand est représenté en vert dans les structures ; le naltrindole est en jaune, et la β-funaltrexamine est en violet. B. Structure chimique du naltrindole et de la leucine-enképhaline avec le groupement chimique conservé (en rouge) constituant une signature moléculaire de la plupart des ligands opiacés. C. Mode de liaison conservé du groupement chimique constituant la signature moléculaire des opiacés et réseau d’interaction avec les récepteurs OR. Le schéma en haut à droite représente une vue globale des récepteurs et indique la position relative de la zone agrandie. |
|
Sélectivité de liaison des OR : le modèle de l’adresse/message La pharmacologie des opiacés a depuis longtemps été décrite par le modèle de l’adresse/message : le ligand est composé de deux modules, l’un comportant l’information relative à la sélectivité (adresse), et l’autre comportant l’information relative à l’activité (message) du ligand (antagoniste ou agoniste). L’étude de la structure des OR montre que cette propriété pharmacologique est la conséquence directe de l’architecture structurale et de la nature des acides aminés formant la poche de liaison des OR. La partie enfouie dans les récepteurs, qui reconnaît le module conférant l’activité des ligands (message), est composée d’acides aminés strictement conservés entre les différents sous-types d’OR (Figure 3A). En revanche, une divergence de séquences existe dans la partie supérieure de la poche de liaison des OR, qui reconnaît le module conférant la sélectivité des ligands (adresse). Elle comporte plusieurs acides aminés responsables de la sélectivité de liaison des ligands (Figure 3B). Par exemple, le groupe indole du naltrindole, qui est responsable de la sélectivité de liaison pour le delta-OR, interagit avec la Leu7.35 dans la partie supérieure de la poche de liaison. Dans le mu-OR, la présence d’un Trp7.35 à cette même position n’est pas favorable à la liaison du naltrindole, avec un conflit stérique entre les deux groupements indole ; ceci constitue la base moléculaire de la sélectivité de liaison du naltrindole pour le delta-OR.
 | Figure 3.
Bases moléculaires et structurales de la sélectivité de liaison des OR. A. Vue de côté de la poche de liaison des OR montrant deux domaines avec une conservation différente de leur séquence primaire. En haut, les séquences non conservées constituant un filtre de sélectivité (domaine adresse) et en bas les séquences strictement conservées qui sont responsables de l’activité des récepteurs (domaine message). β-Fna : β-funaltrexamine. B. Vue de dessus (schéma du haut) et de côté (schéma du bas) du récepteur delta-OR en mode surface présentant les résidus impliqués dans la sélectivité de liaison (en rose). C. La poche de liaison allostérique du récepteur muscarinique M2 (M2R) est formée par des domaines positionnés de façon identique à ceux formant le filtre de sélectivité des OR. |
Ce filtre de sélectivité extracellulaire semble s’appliquer plus largement à d’autres RCPG. Par exemple, pour les récepteurs muscariniques, le domaine adresse constitue le site d’action de modulateurs allostériques (poche allostérique) (Figure 3C). Dans ce cas, par opposition aux OR, il y a une séparation physique entre les deux domaines, et ceci pourrait expliquer la plus grande difficulté à trouver des ligands sélectifs pour les récepteurs muscariniques [
14] (→).
(→) Voir la synthèse de J.L. Galzi et B. Ilien, page 852 de ce numéro
Cette observation peut donc avoir des implications directes dans le développement de ligands sélectifs de sous-types de RCPG en général. |
Oligomérisation du récepteur mu des opiacés Les OR, comme la plupart des RCPG [
9], sont capables de s’associer pour former des dimères ou des oligomères [
10,
15] (→)
(→) Voir la synthèse de J. Kniazeff et J.P. Pin, page 858 de ce numéro
Le rôle de l’oligomérisation dans la fonction des RCPG reste encore très controversé. Pour autant, il semble que la pharmacologie des OR soit modulée par l’existence d’une combinaison mu-OR/delta-OR en complément de celle des récepteurs exprimés individuellement [
11]. La structure du mu-OR révèle la formation d’oligomères à travers deux interfaces d’interaction. La première fait intervenir une partie du TM1, du TM2 et de l’hélice 8 (Figure 4A) avec une surface d’interaction de 615 Å2. L’autre interface, beaucoup plus importante (1492 Å2), est constituée par l’interaction entre le TM5 et le TM6 de chaque récepteur, formant un motif canonique à quatre hélices alpha disposées en faisceau avec un réseau d’interaction faisant intervenir 28 acides aminés (Figure 4A). L’acide aminé Thr en position 279 (Thr6.34), qui joue un rôle clé dans la stabilisation de l’état inactif du récepteur, fait partie de cette interface. Il est donc tentant de spéculer que l’oligomérisation des OR pourrait jouer un rôle important dans la régulation de l’activité du récepteur, et notamment dans son couplage à la protéine G. Il est aussi intéressant de noter que les deux poches de liaison entre deux récepteurs sont couplées par l’intermédiaire d’un réseau d’interactions hydrophobes. Cette observation pourrait donc apporter une explication structurale à l’obtention de profils pharmacologiques différents pour les hétérodimères, et aux phénomènes de coopérativité observés dans les expériences pharmacologiques. Finalement, cette structure ouvre de nouvelles pistes pour étudier le phénomène d’oligomérisation des RCPG et mieux comprendre ses implications fonctionnelles.
 | Figure 4.
Organisation des oligomères de mu-OR. A. Vue de dessus (extracellulaire) montrant les interfaces de contact entre deux mu-OR, l’une impliquant le TM5 et le TM6, et l’autre impliquant les TM1/TM2 et l’hélice 8 (H8). B. Vue de côté des oligomères. C. Surface d’interaction constituée par le TM5 et le TM6 (en bleu). D. Surface d’interaction constituée par les TM1/TM2 et l’hélice 8 (H8) (en bleu). |
|
Les structures décrites dans ces études sont des conformations inactives des OR. Elles permettent de lever un premier voile sur les mécanismes d’action des opiacés. Il est maintenant nécessaire d’obtenir la forme active de ces récepteurs complexés avec les protéines de signalisation, comme les protéines G, et en présence de différents agonistes, comme la morphine et les endorphines. En effet, la morphine déclenche une signalisation cellulaire différente de celle induite par les endorphines, mais les bases moléculaires et structurales qui sous-tendent ces différences sont inconnues. La morphine et les endorphines se liant au même récepteur, ces deux molécules stabiliseraient le mu-OR dans des conformations différentes, à l’origine des différences de réponses biologiques. Une étude récente menée dans notre laboratoire sur un autre RCPG, le récepteur 2 de la vasopressine (V2R), révèle que les RCPG sont en effet capables d’induire des réponses cellulaires différentes en adoptant des conformations distinctes en fonction du ligand lié au récepteur [
12]. La compréhension des bases structurales de l’action de la morphine, et des opiacés en général, pourrait donc permettre de développer des molécules gardant les effets bénéfiques (analgésie) sans pour autant induire d’effets secondaires. Pour conclure, en plus d’apporter de précieuses informations sur le fonctionnement des RCPG en général, la résolution de la structure 3D des OR, clé du traitement de la douleur et des addictions, ouvre la voie à la conception rationnelle de nouveaux ligands, potentiellement plus spécifiques et efficaces. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
|
Ce travail a pu être réalisé grâce au soutien de l’Inserm pour ma mise à disposition auprès de l’université de Stanford. Ces études ont été réalisées avec la participation d’Aashish Manglik, Andrew C. Kruse, Tong Sun Kobilka, Foon Sun Thian, Jesper M. Mathiesen, Roger K. Sunahara, Leonardo Pardo, William I. Weis et Brian K. Kobilka.
|
Footnotes |
1.
Pradhan
AA
,
Befort
K
,
Nozaki
C
, et al.
The delta opioid receptor: an evolving target for the treatment of brain disorders . Trends Pharmacol Sci.
2011; ; 32 : :581.–590. 2.
Satoh
M
,
Minami
M.
Molecular pharmacology of the opioid receptors . Pharmacol Ther.
1995; ; 68 : :343.–364. 3.
Rosenbaum
DM
,
Cherezov
V
,
Hanson
MA
, et al.
GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function . Science.
2007; ; 318 : :1266.–1273. 4.
Granier
S
,
Manglik
A
,
Kruse
AC
, et al.
Structure of the delta-opioid receptor bound to naltrindole . Nature.
2012; ; 485 : :400.–404. 5.
Manglik
A
,
Kruse
AC
,
Kobilka
TS
, et al.
Crystal structure of the micro-opioid receptor bound to a morphinan antagonist . Nature.
2012; ; 485 : :321.–326. 6.
Thompson
AA
,
Liu
W
,
Chun
E
, et al.
Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic . Nature.
2012; ; 485 : :395.–399. 7.
Wu
H
,
Wacker
D
,
Mileni
M
, et al.
Structure of the human kappa-opioid receptor in complex with JDTic . Nature.
2012; ; 485 : :327.–332. 8.
Ballesteros
JA
, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors.
1995 ; San Diego: : Academic Press;
9.
Fanelli
F
,
De Benedetti
PG.
Update 1 of computational modeling approaches to structure-function analysis of G protein-coupled receptors . Chem Rev.
2011; ; 111 : :PR438.–PR535. 10.
Rozenfeld
R
,
Gomes
I
,
Devi
L.
Opioid receptor dimerization . In:
Pasternak
GW
, ed.
The opiate receptors . New York: : Humana Press; , 2011 : :407.–437. 11.
George
SR
,
Fan
T
,
Xie
Z
, et al.
Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties . J Biol Chem.
2000; ; 275 : :26128.–26135. 12.
Rahmeh
R
,
Damian
M
,
Cottet
M
, et al.
Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy . Proc Natl Acad Sci USA.
2012; ; 109 : :6733.–6738. 13.
Lebon
G
,
Tate
C.
Les récepteurs couplés aux protéines G dans la lumière . Med Sci (Paris).
2012; ; 28 : :876.–882. 14.
Galzi
JL
,
Ilien
B.
Les récepteurs couplés aux protéines G : des régulateurs allostériques du métabolisme cellulaire . Med Sci (Paris).
2012; ; 28 : :852.–857. 15.
Kniazeff
J
,
Pin
JP.
Des dimères, des oligomères de RCPG, oui mais pourquoi ? Le récepteur GABAB sous interrogatoire . Med Sci (Paris).
2012; ; 28 : :858.–863. |





