Vignette (Photo © Pixabay - Creative Commons).
| |||
Med Sci (Paris). 33: 27–29. doi: 10.1051/medsci/201733s105.Pneumothorax récidivants chez un patient atteint de dystrophie musculaire congénitale avec déficit en collagène VI 1Hôpital Pierre-Zobda-Quitman, CHU de Martinique, Fort-de-France, Martinique, France 2Hôpital Marin de Hendaye, Centre de Compétence, et FILNEMUS, Marseille, France Corresponding author. | |||
Vignette (Photo © Pixabay - Creative Commons). | |||
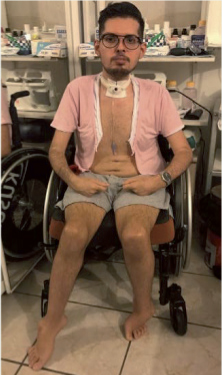
Omar est un jeune homme âgé de 21 ans lors de la survenue de son premier épisode de pneumothorax. Il est originaire de Cuba, né à terme de parents non consanguins sans histoire familiale ni signes évoquant une pathologie neuromusculaire. Dès le 3e trimestre de grossesse avaient été notés un oligoamnios et une diminution des mouvements fœtaux. L’accouchement s’est toutefois déroulé sans incident notable. À la naissance, en revanche, sont observées une hypotonie néonatale et une subluxation de la hanche gauche. Une dysplasie bilatérale des hanches est alors été diagnostiquée. Une ténotomie des adducteurs est réalisée à l’âge de 9 mois, suivie d’une immobilisation par plâtre. Les premières acquisitions motrices ont été retardées car le nourrisson a longtemps porté un appareillage. Il a malgré tout marché à partir de l’âge de 12 mois cependant avec difficultés, sur de courtes distances et de manière anormale (la marche était rapportée comme dandinante). Par la suite, des déformations des pieds se sont installées entraînant un important équin bilatéral responsable d’un arrêt de la marche à partir de l’âge de 5 ans. Une gêne respiratoire nocturne avait été signalée dès cette période mais non évaluée à notre connaissance. Le développement intellectuel était normal. Les taux de CPK étaient peu modifiés lors de l’évolution de la maladie : 97 unités à 2 ans, puis oscillant par la suite autour de 600 unités (3 fois le taux normal), 564 à 3 ans et 626 à 4 ans. Le bilan étiologique réalisé à Cuba, et en premier lieu une biopsie musculaire, a conclu à un tableau de dystrophie mais sans pouvoir étudier les protéines membranaires du muscle. Une étude génétique réalisée en août 1997 n’a pas révélé d’anomalie du gène de la dystrophine. Le patient se rend dans notre service en Martinique à l’âge de 7 ans pour des investigations complémentaires et adaptation de sa prise en charge. L’anamnèse et le tableau clinique permettent de retenir le diagnostic de myopathie à début précoce, possiblement d’étiologie congénitale. L’atteinte motrice est diffuse et importante. La marche est impossible. Le patient parvient à se déplacer à quatre pattes. Les rétractions sont marquées surtout aux membres inférieurs et dans une moindre mesure aux coudes. Il y a une raideur rachidienne et une limitation de flexion du cou. Les doigts et les orteils sont hyperlaxes.
Une nouvelle biopsie musculaire a confirmé l’aspect de dystrophie musculaire et montre dans un premier temps l’absence d’anomalie de la mérosine. L’étude complémentaire du collagène VI sur coupes confirme l’existence d’un déficit partiel (Pr M. Fardeau et Dr N. Romero). Par la suite, une étude génétique familiale viendra confirmer le diagnostic de dystrophie musculaire congénitale de type Ullrich avec déficit en collagène VI. Cette étude est réalisée par le Dr P. Richard en PCR et séquençage direct de type Sanger. Le patient est porteur de deux mutations faux-sens différentes dans le gène COL6A2, l’une héritée du père (c.2809 C>T ; p.Arg937Trp) et l’autre héritée de la mère (c.2192 C>T ; p.Thr731Met). Sur le plan familial, aucun des deux parents ne présente de trouble et deux demi-sœurs et un demi-frère dans la filiation paternelle ne présentent aucun signe de pathologie neuromusculaire. Le bilan pneumologique confirme un important syndrome restrictif avec une capacité pulmonaire totale de 59 % de la valeur théorique. À l’âge de 8 ans, une ventilation non invasive nocturne est mise en place en raison d’un important syndrome restrictif respiratoire. Le suivi montre une stabilisation des paramètres ventilatoires et gazométriques. Un nouveau traitement chirurgical des rétractions des membres inférieurs est réalisé l’âge de 9 ans. Les suites opératoires sont compliquées par un intense tableau algique est une neuro-algodystrophie des articulations des membres inférieurs affectant particulièrement les pieds. De 9 à 21 ans il n’y a pas eu d’évènement significatif. Le déficit moteur s’est très lentement aggravé. Les rétractions segmentaires et axiales se sont progressivement majorées. Les capacités fonctionnelles restaient cependant comparables. L’utilisation d’un fauteuil roulant électrique est devenue nécessaire à partir de l’âge de 13 ans. À partir de l’âge de 21 ans, le patient présente des accès de tachycardie associée à des douleurs thoraciques et une dyspnée. L’ensemble de ces symptômes s’accompagne de malaises, sans perte de connaissance cependant. Après évaluation pneumologique, les paramètres de la VNI sont modifiés et le volume courant augmenté. Le patient présente alors une décompensation de ses troubles avec une aggravation de la dyspnée et des altérations des gaz du sang, en particulier une hypercapnie persistante. Un scanner thoracique est réalisé et révèle un pneumothorax bilatéral qui est pris en charge par drainage. Par la suite quatre nouveaux pneumothorax se produisent dont trois de taille modérée à droite et un plus important à gauche. Face aux récidives des pneumothorax, et à l’importance des troubles engendrés (décompensations respiratoires, douleurs majeures, accès répétitifs de tachycardie), une décision collégiale, impliquant également des collègues de métropole, est prise en vue d’aboutir à un traitement définitif. Le patient est alors âgé de 23 ans. Il a suivi une formation en informatique de gestion mais n’a pu obtenir le diplôme de technicien supérieur. Il n’a pas d’activité professionnelle. Il vit à son domicile, assisté de son épouse et de sa mère. Une symphyse des feuillets pleuraux par talcage est réalisée en deux étapes. La première étape porte sur la cavité droite. Elle est réalisée sans complication et avec des suites simples. Puis la cavité gauche est traitée trois mois après. Des complications surviennent alors avec un hématome pariétal qui nécessitera une nouvelle intervention pour drainage ainsi que des transfusions sanguines. Des complications infectieuses viendront se greffer peu après rendant la situation très instable. Une trachéotomie et une gastrostomie seront alors nécessaires pour passer ce cap difficile. Actuellement, le patient a pu retourner à son domicile. Il est ventilé de façon invasive (trachéotomie) 18 à 20 h par jour. La gastrostomie n’a pas été maintenue et l’alimentation se fait de nouveau par voie orale. Le dernier poids connu était de 41 kg et le BMI à 14,4. Une récidive de pneumothorax s’est produite récemment en juillet 2017. Cela s’est traduit par des douleurs modérées et des accès de tachycardie. Il n’y a pas eu de signe fonctionnel respiratoire. Au scanner, ce pneumothorax est resté limité en taille. Il a évolué favorablement, spontanément. Il y aura eu au total sept pneumothorax en trois ans dont un survenu cinq mois après talcage. | |||
La myopathie d’Ullrich est la forme la plus sévère des dystrophies musculaires congénitales avec déficit en collagène VI. Elle associe un tableau de myopathie rétractile contrastant avec une hyperlaxité distale. L’atteinte respiratoire y est très fréquente, nécessitant dans bon nombre de cas un recours à une assistance respiratoire (invasive ou non). Les déformations rachidiennes relèvent plus du rigid spine que d’une scoliose vraie expliquant qu’il est rare de recourir à une arthrodèse du rachis chez ces patients. La survenue de pneumothorax chez un patient neuromusculaire bénéficiant d’une assistance respiratoire à pression positive n’est pas exceptionnelle. Ceci est généralement lié à des pressions trop élevées délivrées par le ventilateur. Dans le cas présent, le mécanisme pourrait être plus complexe qu’il n’y paraît ; comme le laisse penser la publication très récente de Kristin L. Fraser et al. [1]. Le collagène VI est un élément de structure essentiel du muscle, mais aussi du poumon, et de sa matrice extracellulaire [2]. Il en découlerait une fragilité anormale dans ce cadre, favorisant la survenue de pneumothorax dont certains peuvent être récidivants et multiples [1]. Kristin L. Fraser et al. rapportent deux contextes distincts : un premier groupe est constitué de nouveau-nés ou d’enfants avec un pneumothorax unique consécutifs à un mauvais réglage du ventilateur avec d’importantes modifications de pression intrathoracique. Le second groupe est constitué de patients adultes avec des pneumothorax récidivants associés au scanner à des anomalies du parenchyme pulmonaire. Dans le cas de notre patient, l’affection est apparue en période néonatale, le premier pneumothorax est survenu à l’âge de 21 ans en dehors de tout changement de paramètre de sa VNI. Il est passé inaperçu mais des changements importants de paramètres pourraient avoir favorisé les récidives Le scanner thoracique est l’examen de référence pour dépister ces pneumothorax. Chez notre patient, le premier pneumothorax n’avait pu être détecté que rétrospectivement sur des radios simples. Par la suite, les récidives ont pu être très rapidement identifiées au scanner et prises en charge en conséquence. Ces pneumothorax et les complications consécutives ont conduit chez notre patient à une altération durable des fonctions respiratoires et le passage en ventilation invasive et quasi permanente n’a pu être évité. Les récidives de pneumothorax se sont multipliées après et malgré les drainages réalisés. Le traitement définitif, précoce est donc nécessaire et recommandé dans ce type de situation. Un avis chirurgical précoce est nécessaire. La prise en charge doit être organisée de manière pluridisciplinaire, impliquant les différents spécialistes concernés : myologue, pneumologue, chirurgien thoracique, et réanimateur. L’état général et nutritionnel du patient doit aussi recueillir toutes les attentions et soins nécessaires en période pré et post-opératoire. Une récidive de pneumothorax s’est produite chez notre patient malgré le talcage. Elle est cependant restée limitée, sans incidence sur les paramètres respiratoires, et n’a pas nécessité de drainage.
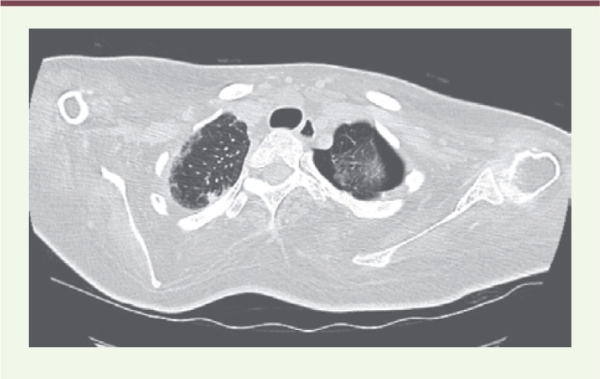
TDM de pneumothorax avant le premier talcage.
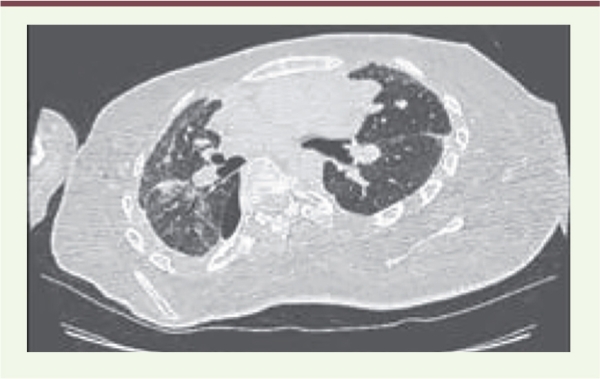
TDM du dernier pneumothorax après les deux talcages. Les conseils de prévention sont également importants à prendre en compte. Il s’agit de l’impact probable de la pression de ventilation mécanique avec une recommandation de limitation des pressions de ventilation au minimum nécessaire. Chez notre patient, l’une des décompensations a probablement été favorisée par une augmentation des paramètres de pression avant que les pneumothorax ne soient détectés. Par ailleurs, les mobilisations brusques ou accidentelles pourraient être des facteurs déclenchant comme lors des actes de kinésithérapie respiratoire ou lors des transferts pour les patients très atteints sur le plan moteur. | |||
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les don- nées publiées dans cet article. | |||