| dc.contributor.author | Fichard, Agnés | - |
| dc.contributor.author | Chanut-Delalande, Hélène | - |
| dc.contributor.author | Ruggiero, Florence | - |
| dc.date.accessioned | 2014-01-09T11:34:48Z | |
| dc.date.available | 2014-01-09T11:34:48Z | |
| dc.date.issued | 2003-04 | fr_FR |
| dc.identifier.citation | Fichard, Agnés ; Chanut-Delalande, Hélène ; Ruggiero, Florence ; Le syndrome d’Ehlers-Danlos : l’architecture matricielle en question, Med Sci (Paris), 2003, Vol. 19, N° 4; p. 443-452 ; DOI : 10.1051/medsci/2003194443 | fr_FR |
| dc.identifier.issn | 1958-5381 | fr_FR |
| dc.identifier.uri | http://hdl.handle.net/10608/4734 | |
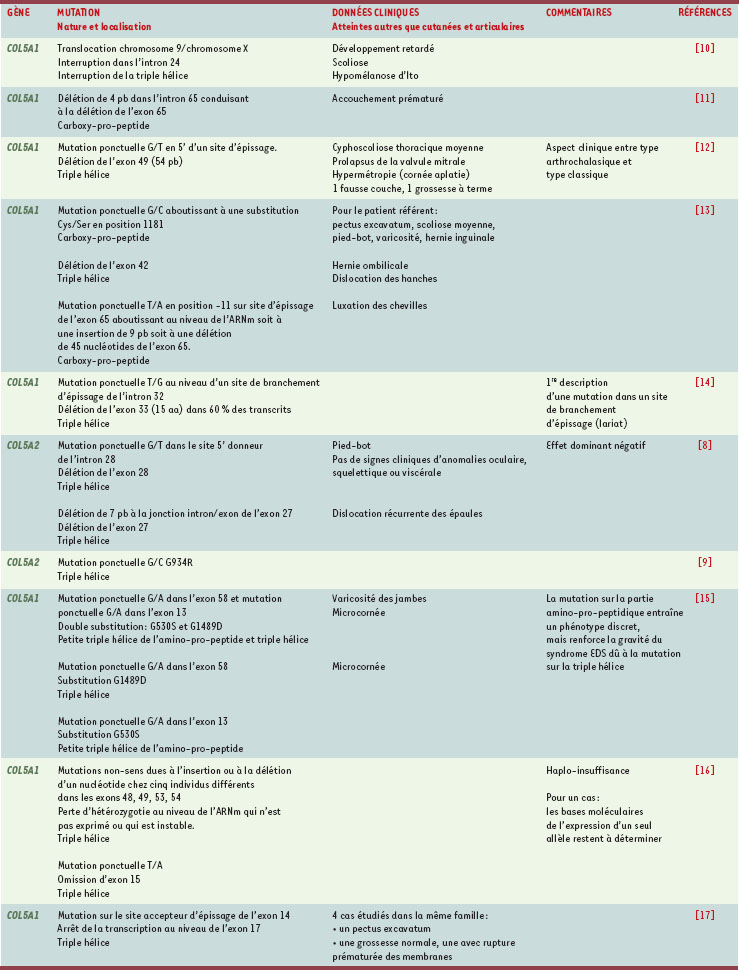
| dc.description.abstract | Le syndrome d’Ehlers-Danlos constitue un groupe hétérogène de maladies génétiques du tissu conjonctif. Il est caractérisé par une peau hyper-extensible, des articulations anormalement mobiles et des vaisseaux fragiles. Les anomalies moléculaires responsables de cette maladie portent souvent sur les collagènes et les enzymes assurant leur maturation. La forme classique du syndrome, qui sera principalement discutée dans cet article, est majoritairement due à des mutations du collagène V, un collagène fibrillaire présent en petite quantité dans les tissus affectés. Cependant, des anomalies moléculaires du collagène I ou de la ténascine peuvent aussi être responsables de ce syndrome. De plus, chez la souris, l’invalidation de gènes codant pour d’autres molécules matricielles (SPARC, thrombospondine, petits protéoglycanes riches en leucine) conduit à des phénotypes mimant ce syndrome et suggère que ces molécules pourraient donc être impliquées. Comme les anomalies du collagène V restent à ce jour principalement responsables de cette affection, nous discuterons son rôle physiologique à la lumière des observations cliniques et fondamentales. Nous tenterons de comprendre comment le collagène V interagit avec les autres molécules pour déterminer les caractéristiques tissulaires. | fr |
| dc.description.abstract | Ehlers-Danlos syndrome (EDS) is a heterogeneous heritable connective tissue disorder characterized by hyperextensible skin, hypermobile joints and fragile vessels. The molecular causes of this disorder are often, although not strictly, related to collagens and to the enzymes that process these proteins. The classical form of the syndrome, which will be principally discussed in this review, can be due to mutations on collagen V, a fibrillar collagen present in small amounts in affected tissues. However, collagen I and tenascin have also been demonstrated to be involved in the same type of EDS. Moreover gene disruption of several other matrix molecules (thrombospondin, SPARC, small leucine rich proteoglycans…) in mice, lead to phenotypes that mimic EDS and these molecules have thus emerged as new players. As collagen V remains the prime candidate, we discuss, based on fundamental and clinical observations, its physiological role. We also explore its potential interactions with other matrix molecules to determine tissue properties. | en |
| dc.language.iso | fr | fr_FR |
| dc.publisher | EDK | fr_FR |
| dc.relation.ispartof | M/S Revues | fr_FR |
| dc.rights | Article en libre accès | fr |
| dc.rights | Médecine/Sciences - Inserm - SRMS | fr |
| dc.source | M/S. Médecine sciences [ISSN papier : 0767-0974 ; ISSN numérique : 1958-5381], 2003, Vol. 19, N° 4; p. 443-452 | fr_FR |
| dc.subject.mesh | Animaux | fr |
| dc.subject.mesh | Collagène | fr |
| dc.subject.mesh | Syndrome d'Ehlers-Danlos | fr |
| dc.subject.mesh | Matrice extracellulaire | fr |
| dc.subject.mesh | Humains | fr |
| dc.subject.mesh | Souris | fr |
| dc.subject.mesh | Mutation | fr |
| dc.subject.mesh | Ténascine | fr |
| dc.title | Le syndrome d’Ehlers-Danlos : l’architecture matricielle en question | fr |
| dc.type | Article | fr_FR |
| dc.contributor.affiliation | Institut de Biologie et Chimie des Protéines, UMR Cnrs 5086, 7, passage du Vercors, 69367 Lyon Cedex 07, France | fr_FR |
| dc.identifier.doi | 10.1051/medsci/2003194443 | fr_FR |
| dc.identifier.pmid | 12836217 | fr_FR |