Hémopathies malignes
2008
| ANALYSE |
16-
Classification
 ). Cette nouvelle classification tient compte du tissu d’origine de la prolifération, lymphoïde ou myéloïde, puis des éléments cliniques, morphologiques ou histologiques, immunophénotypiques, génétiques et moléculaires pour définir chaque entité.
). Cette nouvelle classification tient compte du tissu d’origine de la prolifération, lymphoïde ou myéloïde, puis des éléments cliniques, morphologiques ou histologiques, immunophénotypiques, génétiques et moléculaires pour définir chaque entité.Hémopathies malignes du tissu myéloïde
). À ces deux phénomènes s’ajoutent des anomalies de la différenciation pouvant aller jusqu’au blocage complet. Dans les syndromes myéloprolifératifs on observe une prolifération d’une ou plusieurs lignées myéloïdes sans blocage de différenciation ; dans les syndromes myélodysplasiques, il y a une augmentation de la prolifération cellulaire avec une anomalies de différenciation et un avortement intramédullaire des cellules ; dans les leucémies aiguës myé-loïdes il existe un blocage complet de la différenciation accompagné d’une augmentation de la prolifération cellulaire. Ces éléments expliquent les principaux signes biologiques que l’on observe dans chacune de ces trois catégories : dans les syndromes myéloprolifératifs, une augmentation des cellules sanguines « normales », dans les syndromes myélodysplasiques, une diminution de ces cellules dans le sang contrastant avec un tissu médullaire très riche et enfin dans les leucémies aiguës myéloïdes, un envahissement médullaire et/ou sanguin par des cellules immatures ou blastes (figure 16.1).Tableau 16.I Hémopathies malignes du tissu myéloïde selon les classifications ICD-O-3 (OMS) et proportions relatives chez les adultes selon les données du Registre des hémopathies malignes de Côte d’Or (1980-2002) et chez les enfants selon les données du Registre national des tumeurs de l’enfant
|
ICD-O-3
|
Hémopathies myéloïdes
|
Adulte (%)
|
Enfant (%)
|
|
|
Syndromes myéloprolifératifs chroniques
|
12,1
|
0,9
|
|
9875/3
|
Leucémie myéloïde chronique
| ||
|
9950/3
|
Polyglobulie de Vaquez
| ||
|
9962/3
|
Thrombocytémie essentielle
| ||
|
9961/3
|
Splénomégalie myéloïde
| ||
|
|
Syndromes myélodysplasiques
|
7,3
|
0,7
|
|
9980/3
|
Anémie réfractaire
| ||
|
9982/3
|
Anémie réfractaire avec sidéroblastes en couronne
| ||
|
9983/3
|
Anémie réfractaire avec excès de blastes
| ||
|
9986/3
|
Syndrome 5q -
| ||
|
9985/3
|
Cytopénie réfractaire avec dysplasies multilignées
| ||
|
|
Syndromes myélodysplasiques / myéloprolifératifs
|
3,2
|
1,9
|
|
9945/3
|
Leucémie myélomonocytaire chronique
|
1,0
|
|
|
9946/3
|
Leucémie myélomonocytaire chronique juvénile
|
0,9
|
|
|
9876/3
|
Leucémie myéloïde chronique atypique
| ||
|
|
Leucémies aiguës myéloïdes
|
9,2
|
11,9
|
|
|
Avec anomalies cytogénétiques récurrentes
| ||
|
9896/3
|
t(8;21) (q22;q22), (AML1/ETO)
| ||
|
9866/3
|
t(15;17) (q22;q12), (PML/RARa) (LAM3)
| ||
|
9871/3
|
inv(16) (p13q32) ou t(16;16) (p13;q22) (CBFb/MYH11)
| ||
|
9897/3
|
anomalie du 11q23 (MLL)
| ||
|
9895/3
|
Avec dysplasies multilignées
| ||
|
9920/3
|
Secondaires à des traitements
| ||
|
|
par des agents alkylants
| ||
|
|
par des inhibiteurs de la topoisomérase II
| ||
|
|
Autres
| ||
|
9872/3
|
LAM peu différenciée (M0)
| ||
|
9873/3
|
LAM sans maturation (M1)
| ||
|
9874/3
|
LAM avec maturation (M2)
| ||
|
9867/3
|
LA myélomonocytaire (M4)
| ||
|
9891/3
|
LA monoblastique (M5)
| ||
|
9840/3
|
LA erythroblastique (M6)
| ||
|
9910/3
|
LA mégacaryoblastique (M7)
| ||
|
9870/3
|
LA à basophiles
| ||
|
9931/3
|
Panmyélose aiguë avec myélofibrose
| ||
|
9930/3
|
Sarcome granulocytaire
| ||
|
9805/3
|
LA avec ambiguïté de lignée
|
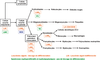
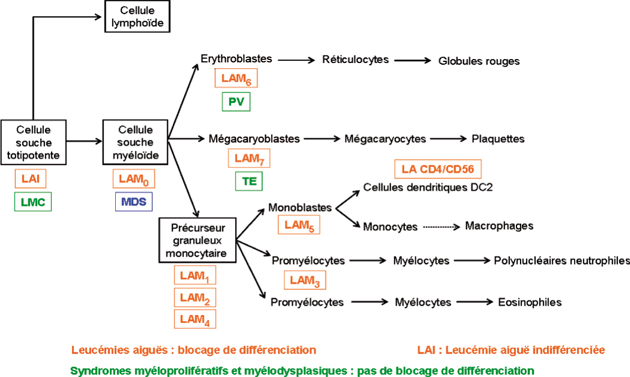
Syndromes myéloprolifératifs chroniques
) juxtapose les gènes Abl du chromosome 9 et Bcr du chromosome 22. Le gène hybride ainsi constitué produit, par l’intermédiaire d’un ARN messager, une protéine anormale qui possède une forte activité tyrosine kinase responsable de modifications de la prolifération cellulaire. Cette fonction de la protéine chimérique est à l’origine des nouveaux outils thérapeutiques doués d’une activité anti tyrosine kinase (Borthakpur et Cortes, 2004). Dans les autres syndromes myéloprolifératifs chroniques, les critères diagnostiques ont toujours été plus délicats à établir avec certitude du fait de possibles évolutions de l’un vers l’autre et de nombreuses formes frontières. La découverte très récente d’une mutation du gène Jak2, également responsable de la synthèse d’une kinase, dans 95 % des polyglobulies de Vaquez et 30 % environ des thrombocythémies essentielles, risque de modifier la classification diagnostique dans l’avenir et ouvre la voie à de nouvelles prises en charge thérapeutiques (James et coll., 2005).Syndromes myélodysplasiques
). Les éléments pronostiques essentiels pris en compte par l’International Prognostic Scoring System (IPSS) dans ces pathologies sont le nombre de cytopénies, le niveau de la blastose médullaire et les anomalies du caryotype (Greenberg et coll., 1997). Au delà de 20 % de blastes médullaires, on considère actuellement qu’il s’agit d’une leucémie aiguë (Jaffe et coll., 2001).Leucémies aiguës myéloïdes
). Le premier élément a été l’abaissement du seuil de blastes à 20 % permettant de les définir ce qui a permis d’intégrer certains syndromes myélodysplasiques particulièrement évolutifs. Le second a été la définition de quatre entités caractérisées par les anomalies cytogénétiques et moléculaires : translocation (15 ; 17), translocation (8 ; 21), inversion du chromosome 16 ou translocation (16 ; 16) et anomalie du 11q2.3 (tableau 16.I). Un troisième élément a été la prise en compte du caractère secondaire à une thérapeutique de la prolifération en raison de leurs circonstances de survenue et de leur pronostic particulièrement péjoratif. Le quatrième nouveau critère est la prise en compte de la notion d’anomalies cytologiques multiples ou dysmyélopoïèse.Hémopathies malignes du tissu lymphoïde
).Tableau 16.II Hémopathies malignes du tissu lymphoïde selon les classifications ICD-O-3 (OMS) et proportions relatives chez les adultes selon les données du Registre des hémopathies malignes de Côte d’Or (1980-2002) et chez les enfants selon les données du Registre national des tumeurs de l’enfant
|
ICD-O-3
|
Hémopathies lymphoïdes
|
Adulte (%)
|
Enfant (%)
|
|
9727-8/3, 9835-6-7/3
|
Leucémies aiguës lymphoblastiques / lymphomes lymphoblastiques
|
1,5
|
57,7
|
|
|
Hémopathies B matures
|
56,6
|
12,6
|
|
9670/3, 9823/3
|
Leucémie lymphoïde chronique/lymphome lymphocytique
|
16,6
| |
|
9833/3
|
Leucémie prolymphocytaire B
| ||
|
9689/3
|
Lymphome splénique à lymphocytes villeux
| ||
|
9940/3
|
Leucémie à tricholeucocytes
|
1,1
| |
|
9732/3
|
Myélome multiple et variant
|
12
| |
|
9762/3
|
Maladies des chaînes lourdes
| ||
|
9699/3
|
Lymphome associé aux muqueuses : MALT extranodal
| ||
|
9699/3
|
Lymphome de la zone marginale nodal
| ||
|
9690-1-5-8/3
|
Lymphome folliculaire
|
5,4
| |
|
9680/3
|
Lymphome diffus à grandes cellules B
|
9,3
|
1,8
|
|
9687/3, 9826/3
|
Lymphome de Burkitt
|
10,7
|
|
|
|
Hémopathies T/NK matures
|
4,0
|
3,0
|
|
9834/3
|
Leucémie prolymphocytaire T
| ||
|
9831/3
|
Leucémie à lymphocytes à grains T
| ||
|
9827/3
|
Leucémie/lymphome T de l’adulte (HTLV1)
| ||
|
9700/3
|
Mycosis fungoïde
|
1,5
| |
|
9701/3
|
Syndrome de Sezary
| ||
|
9718/3
|
Atteintes cutanées primitives CD30+
| ||
|
9708/3
|
Lymphome T sous-cutané de type panniculite
| ||
|
9719/3
|
Lymphome T/NK extra-nodal de type nasal
| ||
|
9709/3
|
Lymphomes T cutanés autres
| ||
|
9714/3
|
Lymphome T ou nul anaplasique à grandes cellules
| ||
|
9650/3 à 9667/3
|
Lymphomes de Hodgkin
|
6
|
12,0
|
|
|
Lymphomes de Hodgkin classiques
| ||
|
|
Lymphome de Hodgkin nodulaire à prédominance lymphocytaire
|
Leucémies aiguës lymphoblastiques
) (Béné et coll., 1995). Ces proliférations sont parfois porteuses d’anomalies cytogénétiques ayant une valeur pronostique importante qui entraîne une prise en charge thérapeutique différente. Les anomalies de bon pronostic sont l’hyperdiploïdie entre 51 et 65 chromosomes et la translocation (12 ; 21) (p13 ; q22) qui juxtapose les gènes TEL et AML-1. Elles sont heureusement celles qui sont le plus largement retrouvées puisque présentes dans plus de la moitié des cas. Au contraire, une hypodiploïdie, une translocation (9 ; 22) (q34 ; q11.2) juxtaposant les gènes BCR et ABL, une translocation (4 ; 11) (q21 ; q23) responsable de la fusion du gène MLL et du gène AF4 ou une translocation (1 ; 19) (q23 ; p13.3) juxtaposant les gènes E2A et PBX sont des anomalies de mauvais pronostic. D’autres anomalies peuvent être retrouvées qui sont associées à un pronostic intermédiaire.
Tableau 16.III Classification immunophénotypique des leucémies aiguës lymphoblastiques selon l’European Group for Immunology of Leukemia (EGIL) (d’après Béné et coll., 1995)
|
Lignée B : toujours C19+ et/ou CD79+ et/ou CD22+
|
|
|
BI (pro-B)
|
Pas d’autre marqueur B
|
|
BII (B commune)
|
CD10+
|
|
BIII (pré-B)
|
Chaîne μ intracytoplasmique
|
|
BIV (B mature)
|
Chaîne légère Kappa ou Lambda en surface
|
|
Lignée T : toujours CD3+ en intracytoplasmique ou en surface
|
|
|
TI (pro-T)
|
CD7+
|
|
TII (pré-T)
|
CD2+ et/ou CD5+ et/ou CD8+
|
|
T-III (T cortical)
|
CD1a+
|
|
T-IV (T mature)
|
CD3+ en surface, CD1a-
|
|
a : α/β T
|
TCR α/β+
|
|
b : γ/δ T
|
TCR γ/δ+
|
Hémopathies lymphoïdes B matures
). À chacune des étapes de cette différenciation caractérisée par des remaniements géniques importants et un fort potentiel prolifératif, des erreurs peuvent se produire qui aboutissent à la prolifération d’un clone lymphoïde malin.) (figure 16.2). L’analyse de l’expression d’un très grand nombre de gènes semble montrer que les LLC sont toutes développées à partir de cellules ayant rencontré l’antigène et donc des cellules mémoire (Klein et coll., 2001; Ghia et coll., 2005). Quoi qu’il en soit, la valeur pronostique du statut mutationnel des gènes des immunoglobulines reste très importante avec un pronostic bien meilleur des formes mutées que des formes non mutées (Vasconcelos et coll., 2003). D’autres marqueurs biologiques comme l’expression du CD38 et celle de ZAP-70 ont été proposées mais n’ont pas encore fait la preuve de leur efficacité ou de leur faisabilité (Crespo et coll., 2003).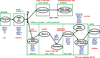 | Figure 16.2 Différenciation du tissu lymphoïde B normal et origine des différentes hémopathies lymphoïdes B |
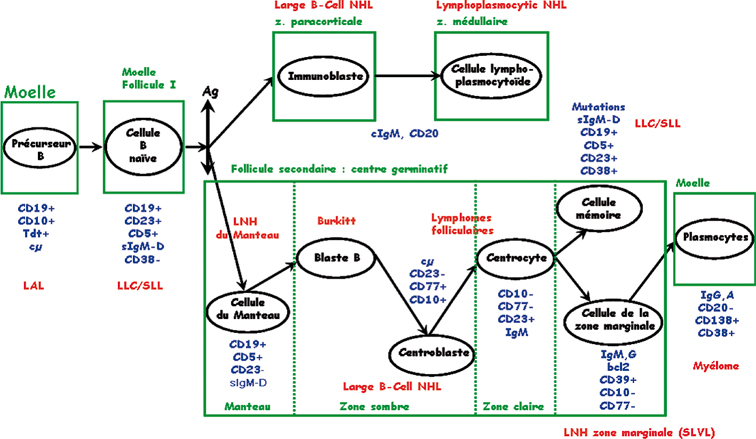 ). De nouvelles prises en charge thérapeutiques ont été décrites ces dernières années utilisant notamment la thalidomide et le bortezomib (Velcade®) (Singhal et coll., 1999) cependant l’affection reste encore d’assez mauvais pronostic avec une survie relative de 42 % à 5 ans (Bossard et coll., 2007).). Ces proliférations sont le plus souvent agressives mais elles ont une chimio-sensibilité assez importante qui leur permet d’avoir un pronostic globalement moins inquiétant. Les éléments pronostiques essentiels sont l’âge, le taux de lacticodéshydrogénase (LDH), le Performance Status, le stade clinique et la localisation extra ganglionnaire regroupés dans un système de score international (Armitage et coll., 1998).). Une trisomie 7 ou 18, une anomalie en 3q27 ou en 6q23-26 ou du 17p peuvent être présentes, les deux dernières étant associées à un mauvais pronostic (Tilly et coll., 1994). Sur le plan génique, il existe des réarrangements du gène BCL2 dans 80 % des cas, des mutations ou des réarrangements de BCL6. Ces lymphomes sont ceux qui ont pour l’instant bénéficié des progrès les plus marquants en terme de prise en charge thérapeutique avec la mise au point d’anticorps humanisés dirigés spécifiquement contre des antigènes exprimés sur les cellules lymphoïdes B comme le CD20 ou le CD52 (Plosker et Figgitt, 2003).). Certaines sont très caractéristiques comme le lymphome de Burkitt dans lequel il existe un aspect cytologique particulier des cellules malignes, un phénotype de cellules B mûres avec expression du CD10, une anomalie cytogénétique et moléculaire impliquant le gène MYC en 8q24 et les gènes des chaînes lourdes ou légères des immunoglobulines en 14q32, en 2q11 ou en 22q11. Cette prolifération de cellules du centre germinatif est très souvent associé au virus d’Epstein-Barr (EBV) qui est retrouvé dans tous les cas endémiques africains mais dans moins de 30 % des cas sporadiques rencontrés dans les autres régions du monde (Anwar et coll., 1995; Tao et coll., 1998). Les lymphomes du manteau sont développés à partir des cellules du manteau du centre germinatif qui ne présentent pas de mutation des zones variables des gènes des immunoglobulines. Leur phénotype immunologique est proche de celui de la leucémie lymphoïde chronique B et on retrouve dans 75 % des cas une translocation (11 ; 14)(q32 ; q32) qui entraîne la juxtaposition des gènes des chaînes lourdes des immunoglobulines et de la cycline D1 responsable d’une hyperexpression de cette molécule (Vandenberghe et coll., 1991).
). De nouvelles prises en charge thérapeutiques ont été décrites ces dernières années utilisant notamment la thalidomide et le bortezomib (Velcade®) (Singhal et coll., 1999) cependant l’affection reste encore d’assez mauvais pronostic avec une survie relative de 42 % à 5 ans (Bossard et coll., 2007).). Ces proliférations sont le plus souvent agressives mais elles ont une chimio-sensibilité assez importante qui leur permet d’avoir un pronostic globalement moins inquiétant. Les éléments pronostiques essentiels sont l’âge, le taux de lacticodéshydrogénase (LDH), le Performance Status, le stade clinique et la localisation extra ganglionnaire regroupés dans un système de score international (Armitage et coll., 1998).). Une trisomie 7 ou 18, une anomalie en 3q27 ou en 6q23-26 ou du 17p peuvent être présentes, les deux dernières étant associées à un mauvais pronostic (Tilly et coll., 1994). Sur le plan génique, il existe des réarrangements du gène BCL2 dans 80 % des cas, des mutations ou des réarrangements de BCL6. Ces lymphomes sont ceux qui ont pour l’instant bénéficié des progrès les plus marquants en terme de prise en charge thérapeutique avec la mise au point d’anticorps humanisés dirigés spécifiquement contre des antigènes exprimés sur les cellules lymphoïdes B comme le CD20 ou le CD52 (Plosker et Figgitt, 2003).). Certaines sont très caractéristiques comme le lymphome de Burkitt dans lequel il existe un aspect cytologique particulier des cellules malignes, un phénotype de cellules B mûres avec expression du CD10, une anomalie cytogénétique et moléculaire impliquant le gène MYC en 8q24 et les gènes des chaînes lourdes ou légères des immunoglobulines en 14q32, en 2q11 ou en 22q11. Cette prolifération de cellules du centre germinatif est très souvent associé au virus d’Epstein-Barr (EBV) qui est retrouvé dans tous les cas endémiques africains mais dans moins de 30 % des cas sporadiques rencontrés dans les autres régions du monde (Anwar et coll., 1995; Tao et coll., 1998). Les lymphomes du manteau sont développés à partir des cellules du manteau du centre germinatif qui ne présentent pas de mutation des zones variables des gènes des immunoglobulines. Leur phénotype immunologique est proche de celui de la leucémie lymphoïde chronique B et on retrouve dans 75 % des cas une translocation (11 ; 14)(q32 ; q32) qui entraîne la juxtaposition des gènes des chaînes lourdes des immunoglobulines et de la cycline D1 responsable d’une hyperexpression de cette molécule (Vandenberghe et coll., 1991).Hémopathies lymphoïdes T
).Lymphomes de Hodgkin
Lymphomes non hodgkiniens
selon leur phénotype B ou T.Bibliographie

→ Aller vers le SOMMAIRE de l'ouvrage