| |
| Med Sci (Paris). 35(3): 223–231. doi: 10.1051/medsci/2019035.Approches nouvelles pour l’étude des interactions
protéine-protéine Benoît Béganton,1 Etienne Coyaud,2 Alain Mangé,1 and Jérôme Solassol1* 1Département de pathologie et oncobiologie, Laboratoire de biologie
des tumeurs solides, CHU de Montpellier, Univ. Montpellier, CHU Arnaud de
Villeneuve, avenue du
Doyen Giraud, 34295Montpellier,
FranceInstitut de Recherche en Cancérologie de Montpellier (IRCM), Inserm
U1194, Univ. Montpellier, 34070Montpellier,
France 2Princess Margaret Cancer Centre, University Health
Network, Toronto, Canada |
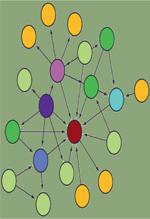
Les interactions protéine-protéine (IPP) jouent un rôle fondamental à tous les niveaux de
la cellule que ce soit dans le métabolisme, la signalisation, la prolifération
cellulaire, la communication intercellulaire ou encore dans le maintien de
l’architecture membranaire. Tous ces processus et fonctions biologiques font intervenir
une multitude de protéines qui agissent de concert dans des systèmes complexes et
interconnectés. Mais la compréhension d’un processus cellulaire, qu’il soit
physiologique ou pathologique, ne se résume plus à la simple identification des
partenaires d’un complexe. Elle doit également permettre d’appréhender les principes
d’association entre protéines au sein des réseaux d’interactions étudiés. L’analyse des
réseaux biologiques et des connexions protéiques directes ou indirectes, transitoires ou
durables, nécessite des approches systémiques complexes à la croisée entre différentes
disciplines comme la biochimie, la protéomique, la biologie cellulaire ou la
bio-informatique. La prise en compte de l’aspect multidimensionnel de l’étude des
réseaux d’interaction, et notamment celui de la dynamique spatio-temporelle, pour
l’identification des IPP et la construction des interactomes qui en résultent a conduit
ces dernières années à des défis technologiques importants. Ainsi, afin de répondre à ce challenge, plusieurs techniques ont été développées
(Figure 1). Parmi elles,
les techniques conventionnelles in vitro, comme la purification par
affinité (ou l’immunoprécipitation) ou le système double hybride chez la levure, bien
que performantes, apparaissent insuffisantes. En effet, ces techniques n’identifient que
les interactions fortes sans tenir compte du contexte cellulaire, ne donnant ainsi qu’un
reflet partiel des IPP. Elles permettent de mettre en évidence des interactions directes
dans des complexes à forte affinité, mais elles ne tiennent pas compte, ou
difficilement, de la dynamique spatio-temporelle. De nombreuses IPP ont toutefois pu
être identifiées par ces techniques, dont beaucoup sont encore référencées dans des
bases de données telles que BioGrid, IntAct, Mint ou DIP.
 | Figure 1. Vue d’ensemble des techniques d’identification des interactions
protéine-protéine. Le réseau d’interaction, au
centre du disque orange, peut être construit au moyen d’approches
conventionnelles (Y2H, AP-MS, FRET, PLA) ou d’approches récentes (APEX,
BioID, Split-BioID). Les techniques d’APEX, BioID et AP-MS sont
principalement utilisées pour caractériser le réseau d’interactions d’une
protéine « A » sans a priori d’interactions. À l’inverse, les techniques de
Y2H, FRET et PLA sont principalement utilisées pour valider une interaction
supposée entre une protéine « A » et une protéine « B ». Les techniques
présentées ne sont pas exhaustives. AP-MS : affinity purification-mass
spectrometry ; APEX : engineered ascorbate peroxidase ; BioID :
proximity-dependent biotinylation identification ; FRET : Förster resonance
energy transfer ; PLA : proximity ligation assay ; Y2H :
yeast-two-hybrid. |
Récemment, de nouvelles approches ont été proposées pour répondre à ces écueils et
permettre d’identifier des interactions in cellulo et in
vivo tout en tenant compte de leurs dimensions spatio-temporelle et
transitoire. Il s’agit de techniques de marquage de proximité par des systèmes
enzymatiques (ou proximity-dependent labeling) comme le BioID
(proximity-dependent biotinylation identification) ou l’APEX
(engineered ascorbate peroxidase). Ces approches ouvrent de
nouvelles perspectives pour l’identification d’interacteurs en contextes physiologique
et pathologique. Elles permettent d’affiner les cartographies des réseaux d’interactions
protéiques au niveau cellulaire et appréhendent plus finement les dérégulations des IPP
que l’on sait être impliquées dans de nombreuses pathologies. Au travers de cette revue, nous nous proposons de présenter les principales technologies
appliquées à l’étude des IPP en nous focalisant plus particulièrement sur les approches
in vivo de type BioID et APEX, lesquelles nous semblent être, à ce
jour, les techniques les plus prometteuses pour l’étude des réseaux d’interactions. |
Approches conventionnelles et exploration des réseaux d’IPP Les purifications par affinité La purification par affinité, et notamment par immuno-affinité, est une des
stratégies les plus couramment utilisées pour identifier les partenaires d’une
protéine d’intérêt. Elle consiste, à partir d’un lysat cellulaire plus ou moins
complexe, en la purification d’une protéine d’intérêt associée à ses partenaires
à l’aide d’un anticorps spécifique [ 1] (Figure
1). Le lysat protéique peut être obtenu à partir de cellules
en culture ou d’extraits tissulaires en utilisant des conditions de lyses douces
et non-dénaturantes essentielles pour préserver au mieux les IPP. Un anticorps
dirigé spécifiquement contre la protéine d’intérêt est ajouté au lysat
cellulaire. Le complexe protéines-anticorps est ensuite purifié à l’aide de
billes de sépharose couplées aux protéines A ou G, lesquelles possèdent une très
forte affinité pour la région Fc des immunoglobulines G (IgG) utilisées. Une
série de lavage avec une stringence plus ou moins forte permet d’éliminer
d’éventuels faux positifs 1. Les complexes
protéiques sont ensuite élués des billes en utilisant différents tampons variant
en fonction des analyses ultérieures : western-blot ou ELISA
pour confirmer des interactions supposées, et spectrométrie de masse pour
identifier plus largement les protéines constituant les complexes purifiés
[ 2]. La grande
popularité de cette technique a conduit à sa large utilisation et à la
publication d’articles majeurs décryptant les réseaux d’interactions
protéine-protéine à l’échelle entière du protéome, et notamment humain
(Tableau I). Ces
articles ont mis à disposition une quantité massive de données utilisées par
l’ensemble de la communauté scientifique et ont été à l’origine d’avancées
majeures des connaissances en biologie [ 3- 5].
Table I.
| Approche |
Complexes protéiques, compartiments ou
fonctions cellulaires ciblés |
Protéine d’intérêt |
Nombre d’interacteurs uniques identifiés |
Nombre moyen d’interacteurs identifiés par
protéine |
Nombre d’interactions identifiées |
Réf |
| AP-MS |
Tout compartiment |
338 |
2 235 |
7 |
6 463 |
[4] |
| Tout compartiment |
1 092 |
11 485 |
11 |
NA |
[51] |
| Tout compartiment |
1 125 |
5 400 |
5 |
28500 |
[3] |
| Tout compartiment |
2 594 |
7 668 |
3 |
23744 |
[6] |
| Tout compartiment |
5 891 |
10961 |
2 |
56553 |
[5] |
|
| Y2H |
Tout compartiment |
7 200 |
1 549 |
NA |
2 754 |
[52] |
| Tout compartiment |
4 456 |
5 632 |
NA |
3 269 |
[53] |
| Signalisation cellulaire |
473 |
1 126 |
NA |
2 626 |
[54] |
| Protéines hépatiques |
5 026 |
3 484 |
NA |
2 582 |
[55] |
| Tout compartiment |
15517 |
4 303 |
NA |
13944 |
[56] |
|
| BioID |
Voie de signalisation Hippo |
19 |
337 |
18 |
487 |
[22] |
| Cils cellulaires |
58 |
1 774 |
31 |
7 092 |
[23] |
| Histone et complexe Mediator |
5 |
452 |
90 |
543 |
[57] |
| Facteur de transcription |
SOX2 |
82 |
82 |
82 |
[24] |
| Transport nucléo-cytoplasmique |
16 |
1 252 |
78 |
ND |
[58] |
| Granules de stress et P-bodies
asso- ciés à l’ARN |
119 |
1 792 |
15 |
7 424 |
[59] |
|
| APEX |
Matrice mitochondriale |
Peptide signal |
495 |
NA |
495 |
[35] |
| Espace intermembranaire mitochon- drial |
Peptide signal |
127 |
NA |
127 |
[40] |
| Jonction RE-MP |
STIM1 |
38 |
38 |
38 |
[60] |
| Cils cellulaires |
NPHP3 |
162 |
162 |
622 |
[61] |
| Granules de stress |
G3BP1 |
123 |
123 |
123 |
[37] |
Sélection d’études d’interactomes. Comparativement
aux techniques d’AP-MS et de Y2H, le BioID et l’APEX permettent
d’identifier plus largement les interacteurs de protéines d’intérêt.
AP-MS : affinity purification-mass spectrometry ;
APEX : engineered ascorbate peroxidase ; BioID :
proximity-dependent biotinylation
identification ; NA : non applicable ; ND : non
disponible ; RE-MP : réticulum endoplasmique-membrane plasmique ;
Y2H : yeast-two-hybrid ; STIM1 : stromal
interaction molecule 1 ; NPHP3 : nephrocystin
3 ; G3BP1 : G3BP stress granule assembly factor
1 ; SOX2 : SRY (sex-determining region Y)-box
2.
|
Cette approche présente un certain nombre d’avantages (Tableau II). Grâce aux
conditions de lyse douce, il est possible de purifier la protéine d’intérêt et
ses interacteurs dans des conditions quasi physiologiques, dans leur
conformation native et en préservant leurs modifications post-traductionnelles.
Par des analyses séquentielles, il est également possible d’étudier les IPP au
cours du temps en réponse, par exemple, à un stimulus. La relative simplicité de
sa mise en œuvre en fait une approche de choix pour caractériser les réseaux
d’interactions protéiques des cellules eucaryotes et pour en révéler toute
l’étendue et la complexité [3, 6]. Parmi les inconvénients des approches de
purification par immuno-affinité, on peut noter qu’elles ne permettent pas, ou
très difficilement, d’étudier des interactions protéiques transitoires ou de
faible affinité. Par ailleurs, les conditions de lyse et de lavage à faible
stringence, nécessaires à la purification des complexes protéiques, présentent
des écueils majeurs parmi lesquels on retrouve l’impossibilité d’étudier ou de
caractériser des protéines faiblement solubles comme les protéines membranaires,
ainsi que l’existence de nombreux faux positifs associés aux complexes purifiés
sans qu’ils en soient spécifiques (Figure
2). Enfin, cette technique nécessite d’utiliser des
anticorps ayant une haute spécificité et une forte affinité pour la protéine
d’intérêt, limitant fréquemment les possibilités d’étude de certaines protéines
pour lesquelles ces anticorps n’existent pas ou n’ont pas ces qualités.
 | Figure 2. Spécificité et résolution des techniques
d’identification des interactions
protéine-protéine. Répartition des techniques
d’AP-MS, APEX, BioID, FRET, PLA et Y2H, selon le nombre
d’interactions identifiées par expérience (résolution) et la
spécificité des interactions protéiques observées. Sont représentées
certaines des techniques d’identification à haut/moyen débit (APEX,
BioID, Split BioID, AP-MS, Y2H) ou à bas débit (Y2H, FRET, PLA). Les
techniques de marquage de proximité (APEX, BioID) sont aujourd’hui
les techniques les plus résolutives et les plus spécifiques. Le
terme BioID utilisé dans la figure correspond au BioID et au
Split-BioID. AP-MS : affinity purification-mass spectrometry ; APEX
: engineered ascorbate peroxidase ; BioID : proximity-dependent
biotinylation identification ; FRET : Förster resonance energy
transfer ; PLA : proximity ligation assay ; Y2H :
yeast-two-hybrid |
Table II.
| Approche |
AP-MS |
APEX |
BioID |
FRET |
PLA |
Y2H |
|
Mécanisme
|
Purification par affinité de complexes
protéiques (principalement par immunoprécipitation) |
Marquage proximal avec de la biotine
activée |
Marquage proximal avec de la biotine
activée |
Émission de fluorescence par tranfert d’énergie
de résonance entre fluorophores |
Détection de fluorescence conjuguée à de l’ADN
amplifié |
Expression d’un gène rapporteur par
complémentarité de deux domaines fonctionnels |
|
Période d’identification
|
À un instant donné |
Quelques minutes |
~24 heures |
À un instant donné |
À un instant donné |
À un instant donné |
|
Avantages
|
Identification d’interactions directes |
Marquage in vivo |
Marquage in vivo |
Identification d’interactions directes |
Facilité technique et rapidité de
l’expérience |
Facilité de culture |
| Possibilité d’étiqueter la protéine cible |
Identification d’interactions fortes, faibles
et transitoires |
Identification d’interactions fortes, faibles
et transitoires |
Identification de l’interaction in
vivo |
Visualisation de la localisation de
l’interaction |
Outils moléculaires à disposition nombreux |
| Peut être quantitative |
Adapté à l’étude de protéines membranaires |
Adapté à l’étude de protéines membranaires |
Visualisation de la localisation de
l’interaction |
Possibilité d’étiqueter les protéines
cibles |
Automatisation des cribles |
|
Inconvénients
|
|
Faux-positifs limités |
Faux-positifs limités |
|
|
|
| Identification d’interactions fortes
seulement |
Nécessite de générer une protéine de
fusion |
Nécessite de générer une protéine de
fusion |
Nécessite de générer des protéines de
fusion |
Utilisation de matériel fixé |
Contexte cellulaire non pertinent |
| Non adapté à l’étude de protéines
membranaires |
Taille de l’enzyme APEX (~28 kDa) |
Taille de l’enzyme BirA* (~32 kDa) |
Taille des fluorophores |
Nécessite des anticorps validés en
immunochimie |
Modifications post-traductionnelles
non-exhaustives |
| Risque de faux-positifs |
Stress oxydatif par le substrat (peroxyde
d’hydrogène) |
La biotinylation peut perturber le comportement
cellulaire |
Calculs de quantification des signaux
d’émission |
Expérience onéreuse |
Taux élevé de faux-positifs |
|
Applicable sur modèles cellulaires ou animaux
|
Oui |
Oui |
Oui |
Oui |
Oui |
NA |
|
Applicable sur échantillons cliniques
|
Oui |
Non |
Non |
Non |
Oui |
NA |
|
Lecture
|
Spectrométrie de masse
Western-blot |
Spectrométrie de masse
Western-blot |
Spectrométrie de masse
Western-blot |
Microscopie |
Microscopie |
Culture par sélection |
Particularités, avantages et inconvénients des techniques
d’identification des IPP. AP-MS : affinity purification-mass
spectrometry ; APEX : engineered ascorbate
peroxidase ; BioID : proximity-dependent
biotinylation identification ; FRET : Förster
resonance energy transfer ; NA : non applicable ; PLA :
proximity ligation assay ; Y2H :
yeast-two-hybrid. |
Pour des cellules en culture, la protéine d’intérêt peut également être étiquetée
avec un peptide (GFP [green fluorescent protein], FLAG-TAG2, etc.), ce qui permet d’utiliser des
anticorps de hautes spécificité et affinité vis-à-vis de ces étiquettes et
d’améliorer les étapes de purification. C’est le principe utilisé par la méthode
TAP (tandem affinity purification) qui utilise un double
étiquetage de la protéine et une double purification d’affinité afin d’augmenter
la spécificité et limiter le taux de faux positifs [7]. D’autres approches, comme le «
pull-down » qui repose sur des protéines « appât »
réactives, permettent de s’affranchir de l’utilisation d’anticorps
spécifiques. Le système de double-hybride chez la levure Développée à la fin des années 1980 [ 8], l’approche double-hybride utilise la
levure Saccharomyces cerevisiae pour explorer des IPP binaires.
Ce système est fondé sur la complémentarité de différents domaines isolés de
facteurs de transcription de la levure pour activer un gène rapporteur et ainsi
produire un signal quantifiable : la protéine d’intérêt est fusionnée à un
domaine de fixation à l’ADN et les protéines cibles à un domaine d’activation de
la transcription. Si les deux protéines interagissent entre elles, le complexe
devient fonctionnel et active la transcription d’un gène rapporteur
(Figure 1). Les
domaines de fixation à l’ADN et d’activation sont généralement issus du facteur
de transcription GAL4. Plusieurs gènes rapporteurs peuvent être utilisés, comme
des gènes contrôlant la croissance de la cellule ou le gène
lacZ codant la β-galactosidase nécessaire au crible
blanc/bleu révélateur de l’IPP 3 [ 9]. Il peut s’agir
d’approches séquentielles évaluant des interactions supposées, l’une après les
autres, ou d’approches matricielles à haut débit. Ces dernières sont fondées sur
le criblage de banques d’ADN complémentaires et permettent de tester plus de
6 000 interactions différentes en un nombre réduit d’expériences [ 10, 11]. L’ensemble de ces méthodes a
permis des analyses à l’échelle du protéome, en particulier humain, et a révélé,
comme pour les purifications d’affinité, de larges réseaux d’interactions
incluant plusieurs centaines, voire parfois plusieurs milliers de partenaires
(Tableau I).
D’autres études, à une échelle plus réduite, ont permis d’identifier des
mécanismes biologiques de maladies neurodégénératives comme la maladie de
Huntington [ 12], ou des
interacteurs de protéines surexprimées dans plusieurs cancers [ 13]. Le système
double-hybride a donc été très largement utilisé ces
dernières années et a permis de déterminer un grand nombre d’interactions
référencées dans les bases de données. Cependant, il présente un certain nombre
d’inconvénients (Tableau
II). S’il permet, en effet, d’identifier des IPP binaires
permanentes, il révèle difficilement les interactions transitoires et, à
l’inverse, un trop grand nombre d’interactions qui ne sont pas physiologiques
(avec des taux de faux positifs estimés entre 25 et 45 %) [ 14] (Figure 2). De plus, plusieurs interactions ne
sont pas identifiées par cette technique en raison de problèmes liés à
l’apparition d’une toxicité, à l’altération de la structure des protéines
fusionnées à GAL4, aux modifications post-traductionnelles propres aux levures
qui sont parfois inadaptées aux protéines étudiées, ou encore à la localisation
particulière d’un des partenaires (protéines membranaires). Plusieurs études
pointent également la faible reproductibilité des résultats obtenus lors
d’analyses indépendantes [ 9]. Des
évolutions méthodologiques ont toutefois permis de lever certaines limites,
comme la méthode yeast-3-hybrid qui détermine les interactions
entre 3 protéines [ 15],
ou la méthode MYTH ( membrane yeast two-hybrid) qui utilise la
complémentarité de l’ubiquitine pour étudier l’interaction entre deux protéines
membranaires [ 16]. Pour
une vision plus exhaustive des méthodes d’identification des interactions
protéine-protéine chez la levure, le lecteur pourra se référer à la revue de
Anna Brückner et al. [ 17]. |
Exploration des réseaux d’IPP par marquage de proximité : une nouvelle
vision Comme nous l’avons vu, la plupart des méthodes développées pour étudier les IPP sont
limitées à l’identification d’interactions fortes, essentiellement permanentes et à
un instant donné et, parfois, dans un contexte cellulaire non adapté. Elles sont
également peu ou pas appropriées pour l’analyse de protéines membranaires. Deux
techniques récentes d’analyse in vivo des IPP permettent de lever
ces limitations : les techniques de marquage de proximité BioID et APEX. Le marquage
de proximité repose sur la fusion d’une protéine d’intérêt à une enzyme, la biotine
ligase BirA pour le BioID, et l’ascorbate peroxydase pour l’APEX. Lorsqu’elles sont
actives, ces enzymes génèrent un produit réactif qui diffuse autour de la protéine
d’intérêt à laquelle elles sont liées et des protéines environnantes. Il induit
ainsi un marquage de proximité covalent qui permet de les purifier et de les
identifier (Figure 1) (pour
revue, voir [18, 19]). Le BioID : proximity-dependent biotinylation identification Développée en 2012, le BioID utilise la capacité naturelle de l’enzyme BirA à
biotinyler les protéines [ 20]. Cette enzyme bactérienne, isolée d’ Escherichia
coli, est un co-facteur de l’acétyl-CoA carboxylase. Elle a pour
fonction de biotinyler une de ses sous-unités et de réguler l’opéron biotine.
Dans l’approche BioID, et en présence d’ATP et de biotine, BirA va générer
l’espèce réactive biotinoyl-5’-AMP (biotin-AMP), la retenir dans son site actif,
et faciliter son transfert sur un résidu lysine d’une protéine cible. Afin de
permettre un marquage plus large des protéines à proximité, un variant de BirA
(BirA*, portant la mutation p.R118G) est utilisé. BirA* a une affinité plus
faible pour la biotin-AMP que BirA, ce qui permet la libération du site actif et
la biotinylation des protéines environnantes dans un rayon de 10 nm [ 21]. Ce marquage est en
général effectué sur une durée de 24 heures, permettant de retranscrire la
dynamique des interactions protéiques, notamment les interactions transitoires,
dans des conditions physiologiques, et avec une bonne fiabilité (Figure 2). Un des autres
avantages du BioID est de pouvoir utiliser des conditions stringentes de lyse et
de lavage lors de la purification par affinité, permettant ainsi de solubiliser
les protéines membranaires, de les identifier, d’augmenter la pureté des
échantillons et limiter le taux de faux positifs, et de mettre en évidence les
IPP faibles et transitoires, ce qui est habituellement impossible avec les
techniques classiques (Tableau
II). Cette approche a ainsi permis de caractériser à large
échelle l’interactome de la voie de signalisation Hippo, qui oscille rapidement
d’un état inactif à actif en réponse aux stimulus de l’environnement cellulaire
[ 22], ou de cartographier le réseau
d’interactions de plus de 58 protéines localisées à l’interface des centrosomes
et des cils dans les cellules humaines [ 23]. En cancérologie, et plus particulièrement dans le cas des
carcinomes pulmonaires épidermoïdes, le BioID a, pour la première fois, permis
de révéler l’implication d’EP300 ( E1A-binding protein p300),
une histone acétyl-transférase, comme interacteur spécifique du facteur de
transcription SOX2 ( SRY[sex-determining region Y]-box 2) et son
rôle clef dans la différenciation et la croissance tumorale [ 24] (Tableau I). En 2015, le BioID a aussi permis,
en utilisant des xénogreffes de cellules humaines, d’identifier de nouveaux
interacteurs de c-Myc, un proto-oncogène découvert il y a presque 40 ans et dont
la compréhension reste toujours incomplète [ 25, 26]. Dans le domaine des maladies infectieuses, le BioID a
été utilisé avec succès dans l’étude de la transmission à l’homme du parasite
Plasmodium berghei au travers de l’identification de MTRAP
( merozoite-specific thrombospondin-related anonymous
protein), une protéine libérée par le parasite lors du processus
d’invasion et qui interagit avec l’aldolase de l’hôte [ 27]. Enfin, le BioID a été transposé avec
succès en biologie végétale, notamment par l’étude du facteur de transcription
OsFD2 (impliqué dans le développement des feuilles) dans les protoplastes de riz
[ 28]. Il fait ainsi
désormais partie des outils d’étude des interactions protéine-protéine pour les
botanistes [ 29, 30]. Bien que cette approche présente de nets avantages par rapport aux approches
classiques, on peut néanmoins noter qu’elle est associée à quelques
inconvénients. Ainsi, elle nécessite la présence et l’accès de lysines à la
surface des protéines d’intérêt pour en permettre la biotinylation par BirA*.
Par ailleurs, BirA* est une protéine de taille relativement importante par
rapport aux étiquettes plus classiques comme l’hémagglutinine (HA) ou FLAG-TAG
(32 kDa versus 1 kDa). Cette contrainte peut altérer la
structure de la protéine cible et/ou en modifier le comportement ou l’activité.
Les biotinylations des lysines sur la protéine cible peuvent également altérer
son comportement vis-à-vis de ses interacteurs (Tableau II). Précisons que le temps nécessaire
à la biotinylation des protéines est relativement long (environ 24 heures), ce
qui n’est pas adapté à l’identification d’interactions protéiques à cinétique
rapide. Ainsi, des biotine ligases de tailles plus réduites et à activités
enzymatiques plus élevées continuent d’être recherchées ou développées afin
d’améliorer la caractérisation des réseaux d’interactions protéiques. Grâce à un
temps de marquage réduit (environ 10 min), le TurboID présente ainsi un avantage
significatif comparé au BioID pour donner une résolution précise des
interactomes [31]. Un écueil généralement attribué aux techniques de marquages de proximité est le
risque d’identifier des protéines proches de la protéine d’intérêt, mais qui
sont en fait en dehors de son réseau d’interaction. Les approches combinées de
BioID et de purification d’affinité permettent alors de remédier à cet écueil,
identifiant, en une seule expérience, les interactions directes et proximales de
la protéine d’intérêt [32]. Au final, le BioID offre une nouvelle approche pour l’analyse des réseaux
d’interactions et permet de mettre en évidence des interactions de proximité
décrivant l’environnement local dans lequel ces interactions se réalisent. Le split BioID Une variante conditionnelle du BioID a été proposée très récemment [ 33]. Dans cette approche,
deux domaines fonctionnels de BirA* sont fusionnés à deux interacteurs
d’intérêt. Lorsque ces deux protéines se rencontrent, les deux domaines de BirA*
interagissent également, rendant l’enzyme fonctionnelle. Ce n’est donc que
lorsque le complexe protéique est formé que la biotine-AMP sera générée et
pourra biotinyler les interacteurs du complexe étudié (Figure 1) [ 33]. Cette approche permet donc de réduire
significativement le spectre des IPP pour ne s’intéresser qu’aux interacteurs
spécifiques du complexe protéique étudié [ 33, 34]. Le marquage par l’ascorbate peroxydase (APEX) La méthode APEX, développée en 2013, repose sur le même principe que le BioID
[ 35]. Elle utilise une enzyme
différente, l’ascorbate peroxydase monomérique, pour biotinyler les protéines
présentes à proximité d’une protéine d’intérêt (Figure 1). En présence de peroxyde d’hydrogène,
cette enzyme oxyde le biotine-phénol en biotine-phénoxyl, un radical à demi-vie
courte (moins de 2,5 ms) qui va se lier de façon covalente avec les acides
aminés riches en électrons, tels que les tyrosines, les tryptophanes, les
histidines et les cystéines, et ceci dans un rayon d’environ 20 nm autour de la
protéine cible. La principale différence avec le BioID réside dans la durée
d’incubation nécessaire pour biotinyler les protéines, beaucoup plus courte avec
le peroxyde d’hydrogène (entre une minute à quelques heures) du fait de la
toxicité du composé sur les cellules. Comme pour BirA, un variant de l’APEX
(APEX2, p.A134P) a été développé pour augmenter l’efficacité catalytique de
l’enzyme et permettre des études sur des niveaux d’expression plus faibles de la
protéine d’intérêt [ 36]. Si l’APEX propose une description rétrospective des interactions
protéiques un peu moins large que ne le fait le BioID, cette technique est
néanmoins particulièrement adaptée à l’identification spécifique des
interactions protéiques transitoires dans une fenêtre de temps courte
( Figure 2 et
Tableau II).
C’est le cas, par exemple, des réseaux d’interactions de protéines de
signalisation cellulaire dont l’activation est très rapide. L’approche APEX a
été utilisée dans quelques applications cliniques, dont une étude sur les
granules de stress, des agrégats de ribonucléoprotéines impliquées notamment
dans des maladies neurodégénératives comme la maladie d’Alzheimer [ 37], ou une étude sur le protéome des
gouttelettes lipidiques retrouvées dans des maladies du métabolisme comme le
diabète [ 38]. Dans les
deux cas, des réseaux protéiques denses jusqu’alors inconnus ont pu être mis en
évidence, ouvrant de nouvelles voies dans la compréhension de ces maladies et
l’identification de nouvelles cibles thérapeutiques. L’APEX, comme le BioID, sont des techniques transposables chez des organismes
multicellulaires comme la souris et la drosophile, ce qui marque un pas de plus
dans la caractérisation des interactions protéiques dans un contexte in
vivo pertinent [25, 39]. Au-delà de l’étude
des réseaux d’interactions, l’approche APEX permet également de réaliser des
cartographies protéiques d’organelles ou de compartiments subcellulaires, comme
la matrice mitochondriale ou l’espace inter-membranaire mitochondrial
(Tableau I)
[35, 40]. Mais comme le BioID, l’APEX présente un certain nombre
d’inconvénients. Elle nécessite notamment, comme pour BirA, une accessibilité
des acides aminés pour un marquage efficace des complexes protéiques.
L’exposition au peroxyde d’hydrogène peut également modifier la physiologie de
la cellule en activant des voies de signalisation en réponse au stress oxydatif,
ou la dynamique des organelles, ou les interactions protéiques dans leur
ensemble (Tableau
II). Après leur isolement, l’identification des interacteurs pourra être réalisée par
spectrométrie de masse. Si cette méthode fournit une vision plus exhaustive que
les techniques plus traditionnelles comme le western blot, elle
induit, en elle-même, plus de complexité dans les analyses. Plusieurs méthodes
d’analyses statistiques des données recueillies sont en effet proposées ; elles
impactent directement le nombre d’interacteurs qui seront identifiés [41, 42]. Il est donc finalement
nécessaire de s’interroger sur la méthode de sélection des intéracteurs, selon
la question biologique que l’on se pose. |
Les méthodes in situ de validation des interactions protéiques Lorsqu’une interaction protéique est révélée, elle nécessite généralement d’être
confirmée par une technique différente. Des méthodes à plus faible débit ont donc
été développées, comme le PLA (proximity ligation assay) et les
techniques de transfert d’énergie de résonance, comme le FRET (Förster
resonance energy transfer) ou le BRET (bioluminescence
resonance energy transfer). Ces approches permettent d’évaluer
l’interaction entre deux candidats par l’émission d’un signal lumineux lorsque
l’interaction protéine-protéine est effective (Figure 1). Le PLA (proximity ligation assay) Le PLA, parfois appelé in situ PLA (isPLA), est une méthode de
détection des interactions entre deux protéines par fluorescence [ 43]. Les deux protéines
cibles sont détectées par des anticorps spécifiques, qui seront révélés par des
anticorps secondaires (des anticorps anti-anticorps) conjugués à des
oligonucléotides. Si les deux protéines sont dans un environnement proche (moins
de 40 nm), les oligonucléotides respectifs pourront s’hybrider et former un brin
d’ADN circulaire qui, après ligation, sera amplifié par PCR ( polymerase
chain reaction) en cercle roulant, générant un ADN simple brin
composé de copies répétées. La présence de nucléotides préalablement marqués par
des fluorophores permettra la détection de l’ADN amplifié par microscopie de
fluorescence (Figure 1)
[ 44]. Cette
technique est adaptée à la détection de tous types d’interactions protéiques
impliquant notamment des protéines membranaires, nucléaires ou cytosoliques
[ 45, 46]. Elle peut être
adaptée à différents types d’échantillons, qu’il s’agisse de cellules fixées ou
de coupes de tissus fixés ou congelés (Tableau II). Cette approche a ainsi permis de
quantifier l’interaction entre l’EGFR, le récepteur de l’EGF ( epidermal
growth factor), et la protéine adaptatrice GRB2 ( growth
factor receptor-bound protein 2), et d’utiliser cette mesure comme
marqueur prédictif de la survie globale de patients présentant un adénocarcinome
pulmonaire et traités par les inhibiteurs de tyrosine kinase de l’EGFR de
première génération, indépendamment du statut mutationnel du gène
EGFR [ 47]. Les techniques de transfert d’énergie de résonance Comme le PLA, la technique FRET permet de détecter in vivo les
interactions directes entre deux protéines par émission de fluorescence. Cette
approche nécessite cependant une ingénierie moléculaire afin de faire exprimer
par les cellules étudiées, les protéines d’intérêts fusionnées à un fluorophore
donneur (pour l’une) et un fluorophore accepteur (pour l’autre). Lorsque les
deux protéines fusionnées, qui sont exprimées dans les cellules, ont interagi,
le fluorophore donneur est alors excité par un stimulus lumineux ; il transfère
son énergie par résonnance au fluorophore accepteur, lequel émet à son tour un
signal fluorescent à une longueur d’onde spécifique [ 48] (Figure 1). L’avantage de cette technique est de
permettre une analyse dynamique des interactions in vivo sur
des cellules en culture. La spécificité du transfert d’énergie entre donneur et
accepteur réduit également significativement le taux de faux positifs
(Figure 2).
Néanmoins, une des limitations de cette approche est qu’elle n’est pas
applicable à des échantillons cliniques que l’on ne peut transfecter afin
d’exprimer les protéines fusionnées, comme des coupes de tissus (Tableau II). Le BRET exploite le même principe de transfert d’énergie de résonance que le FRET
à la différence qu’il ne nécessite pas de source extérieure de lumière [49]. Le signal lumineux
d’excitation est, dans ce cas, produit par une enzyme, la luciférase par
exemple, qui émet de la lumière lorsqu’elle est en contact avec son substrat.
Ceci permet un transfert d’énergie dans un rayon réduit d’environ 10 nm. Le BRET
remédie à certains écueils du FRET, comme l’auto-fluorescence, la perte de
fluorescence par photoblanchiment (photobleaching) et la
diffusion dynamique de la lumière (light scatterring) [50]. |
Malgré des contraintes techniques évidentes, les purifications d’affinité ont
longtemps constitué les approches de référence pour la caractérisation des réseaux
d’interactions protéiques. Les outils d’étude des IPP continuent d’évoluer et,
aujourd’hui, nous disposons d’approches permettant des analyses fines de ces
réseaux. Le développement récent des marquages de proximité offre désormais la
possibilité de concevoir de façon radicalement nouvelle les réseaux d’interactions,
avec une cartographie dynamique de plus en plus précise, et ce, dans des domaines
qui ont été jusqu’à maintenant seulement explorés de façon séquentielle. Ce nouveau
regard porté sur les interactions protéine-protéine permet d’envisager un large
champ d’applications, allant de l’identification de cibles thérapeutiques ou de
biomarqueurs, jusqu’à l’exploration fonctionnelle de mécanismes biologiques. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les
données publiées dans cet article.
|
Footnotes |
1. Urh
M,
Simpson
D,
Zhao
K. Affinity
chromatography: general methods . Methods
Enzymol.
2009; ; 463 :
:417.–438. 2. ten Have
S,
Boulon
S,
Ahmad
Y,
Lamond
AI. Mass
spectrometry-based immuno-precipitation proteomics - the user’s
guide . Proteomics.
2011; ; 11 :
:1153.–1159. 3. Hein
MY,
Hubner
NC,
Poser
I
et al.
A human interactome in three quantitative dimensions organized by
stoichiometries and abundances . Cell.
2015; ; 163 :
:712.–723. 4. Ewing
RM,
Chu
P,
Elisma
F
et al.
Large-scale mapping of human protein-protein interactions by mass
spectrometry . Mol Syst Biol.
2007; ; 3 : :89.. 5. Huttlin
EL,
Bruckner
RJ,
Paulo
JA
et al.
Architecture of the human interactome defines protein communities
and disease networks . Nature.
2017; ; 545 :
:505.–509. 6. Huttlin
EL,
Ting
L,
Bruckner
RJ
et al.
The BioPlex network: a systematic exploration of the human
interactome . Cell.
2015; ; 162 :
:425.–440. 7. Gerace
E,
Moazed
D. Affinity
purification of protein complexes using TAP tags .
Methods Enzymol.
2015; ; 559 :
:37.–52. 8. Fields
S,
Song
O. A novel genetic
system to detect protein-protein interactions .
Nature.
1989; ; 340 :
:245.–246. 9. Hamdi
A,
Colas
P. Yeast two-hybrid
methods and their applications in drug discovery .
Trends Pharmacol Sci.
2012; ; 33 :
:109.–118. 10. Uetz
P,
Giot
L,
Cagney
G
et al.
A comprehensive analysis of protein-protein interactions in
Saccharomyces cerevisiae . Nature.
2000; ; 403 :
:623.–627. 11. Ito
T,
Chiba
T,
Ozawa
R
et al.
A comprehensive two-hybrid analysis to explore the yeast protein
interactome . Proc Natl Acad Sci USA.
2001; ; 98 :
:4569.–4574. 12. Tourette
C,
Li
B,
Bell
R
et al.
A large scale Huntingtin protein interaction network implicates
Rho GTPase signaling pathways in Huntington disease .
J Biol Chem.
2014; ; 289 :
:6709.–6726. 13. Shahheydari
H,
Frost
S,
Smith
BJ
et al.
Identification of PLP2 and RAB5C as novel TPD52 binding partners
through yeast two-hybrid screening . Mol Biol
Rep.
2014; ; 41 :
:4565.–4572. 14. Huang
H,
Jedynak
BM,
Bader
JS. Where have all
the interactions gone? Estimating the coverage of two-hybrid protein
interaction maps . PLoS Comput Biol.
2007; ; 3 : :e214.. 15. Zhang
J,
Lautar
S. A Yeast
three-hybrid method to clone ternary protein complex
components . Anal Biochem.
1996; ; 242 :
:68.–72. 16. Johnsson
N,
Varshavsky
A. Split ubiquitin
as a sensor of protein interactions in vivo . Proc
Natl Acad Sci USA.
1994; ; 91 :
:10340.–10344. 17. Brückner
A,
Polge
C,
Lentze
N
et al.
Yeast two-hybrid, a powerful tool for systems
biology . Int J Mol Sci.
2009; ; 10 :
:2763.–2788. 18. Gingras
A-C,
Abe
KT,
Raught
B. Getting to know
the neighborhood: using proximity-dependent biotinylation to characterize
protein complexes and map organelles . Curr Opin Chem
Biol.
2018; ; 48 :
:44.–54. 19. Kim
DI,
Roux
KJ. Filling the
void: proximity-based labeling of proteins in living cells .
Trends Cell Biol.
2016; ; 26 :
:804.–817. 20. Roux
KJ,
Kim
DI,
Raida
M
et al.
A promiscuous biotin ligase fusion protein identifies proximal
and interacting proteins in mammalian cells . J Cell
Biol.
2012; ; 196 :
:801.–810. 21. Kim
DI,
Kc
B,
Zhu
W
et al.
Probing nuclear pore complex architecture with
proximity-dependent biotinylation . Proc Natl Acad
Sci USA.
2014; ; 111 :
:E2453.–E2461. 22. Couzens
AL,
Knight
JDR,
Kean
MJ, et al.
Protein interaction network of the mammalian Hippo pathway
reveals mechanisms of kinase-phosphatase interactions .
Sci Signal.
2013;; 6 : :rs15.. 23. Gupta
GD,
Coyaud
E,
Gonçalves
J
et al.
A Dynamic protein interaction landscape of the human
centrosome-cilium interface . Cell.
2015; ; 163 :
:1484.–1499. 24. Kim
BR,
Coyaud
E,
Laurent
EMN
et al.
Identification of the SOX2 interactome by BioID reveals EP300 as
a mediator of SOX2-dependent squamous differentiation and lung squamous cell
carcinoma growth . Mol Cell Proteomics.
2017; ; 16 :
:1864.–1888. 25. Dingar
D,
Kalkat
M,
Chan
PK
et al.
BioID identifies novel c-MYC interacting partners in cultured
cells and xenograft tumors . J Proteomics.
2015; ; 118 :
:95.–111. 26. Meyer
N,
Penn
LZ. Reflecting on 25
years with MYC . Nat Rev Cancer.
2008; ; 8 :
:976.–990. 27. Kehrer
J,
Frischknecht
F,
Mair
GR. Proteomic
analysis of the Plasmodium berghei gametocyte egressome and vesicular bioID
of osmiophilic body proteins identifies merozoite TRAP-like protein (MTRAP)
as an essential factor for parasite transmission .
Mol Cell Proteomics MCP.
2016; ; 15 :
:2852.–2862. 28. Lin
Q,
Zhou
Z,
Luo
W
et al.
Screening of proximal and interacting proteins in rice
protoplasts by proximity-dependent biotinylation .
Front Plant Sci.
2017; ; 8 : :749.. 29. Lampugnani
ER,
Wink
RH,
Persson
S
et al.
The toolbox to study protein-protein interactions in
plants . Crit Rev Plant Sci.
2018 ; :1.–27. 30. Khan
M,
Youn
JY,
Gingras
AC
et al.
In planta proximity dependent biotin identification
(BioID) . Sci Rep.
2018; ; 8 : :9212.. 31. Branon
TC,
Bosch
JA,
Sanchez
AD
et al.
Efficient proximity labeling in living cells and organisms with
TurboID . Nat Biotechnol.
2018; ; 36 :
:880.–887. 32. Liu
X,
Salokas
K,
Tamene
F
et al.
An AP-MS- and BioID-compatible MAC-tag enables comprehensive
mapping of protein interactions and subcellular
localizations . Nat Commun.
2018; ; 9 : :1188.. 33. Schopp
IM, Amaya
Ramirez
CC,
Debeljak
J
et al.
Split-BioID a conditional proteomics approach to monitor the
composition of spatiotemporally defined protein complexes .
Nat Commun.
2017; ; 8 : :15690.. 34. De Munter
S,
Görnemann
J,
Derua
R
et al.
Split-BioID: a proximity biotinylation assay for
dimerization-dependent protein interactions . FEBS
Lett.
2017; ; 591 :
:415.–424. 35. Rhee
H-W,
Zou
P,
Udeshi
ND
et al.
Proteomic mapping of mitochondria in living cells via spatially
restricted enzymatic tagging . Science.
2013; ; 339 :
:1328.–1331. 36. Lam
SS,
Martell
JD,
Kamer
KJ
et al.
Directed evolution of APEX2 for electron microscopy and proximity
labeling . Nat Methods.
2015; ; 12 :
:51.–54. 37. Markmiller
S,
Soltanieh
S,
Server
KL
et al.
Context-dependent and disease-specific diversity in protein
interactions within stress granules . Cell.
2018; ; 172 : :590.–04
e13.. 38. Bersuker
K,
Peterson
CWH,
To
M
et al.
A Proximity labeling strategy provides insights into the
composition and dynamics of lipid droplet proteomes .
Dev Cell.
2018; ; 44 : :97.–112
e7.. 39. Chen
CL,
Hu
Y,
Udeshi
ND
et al.
Proteomic mapping in live Drosophila tissues using an engineered
ascorbate peroxidase . Proc Natl Acad Sci
USA.
2015; ; 112 :
:12093.–12098. 40. Hung
V,
Zou
P,
Rhee
H-W
et al.
Proteomic mapping of the human mitochondrial intermembrane space
in live cells via ratiometric APEX tagging . Mol
Cell.
2014; ; 55 :
:332.–341. 41. Tyanova
S,
Temu
T,
Cox
J. The MaxQuant
computational platform for mass spectrometry-based shotgun
proteomics . Nat Protoc.
2016; ; 11 :
:2301.–2319. 42. Choi
H,
Larsen
B,
Lin
ZY
et al.
SAINT: probabilistic scoring of affinity purification-mass
spectrometry data . Nat Methods.
2011; ; 8 :
:70.–73. 43. Söderberg
O,
Gullberg
M,
Jarvius
M
et al.
Direct observation of individual endogenous protein complexes in
situ by proximity ligation . Nat Methods.
2006; ; 3 : (995–1)
:000.. 44. Söderberg
O,
Leuchowius
K-J,
Gullberg
M
et al.
Characterizing proteins and their interactions in cells and
tissues using the in situ proximity ligation assay .
Methods San Diego Calif.
2008; ; 45 :
:227.–232. 45. Bobrich
MA,
Schwabe
SA,
Brobeil
A
et al.
PTPIP51: a new interaction partner of the insulin receptor and
PKA in adipose tissue . J Obes.
2013; ; 2013 :
:476240.. 46. Poulard
C,
Treilleux
I,
Lavergne
E
et al.
Activation of rapid oestrogen signalling in aggressive human
breast cancers . EMBO Mol Med.
2012; ; 4 :
:1200.–1213. 47. Smith
MA,
Hall
R,
Fisher
K, et al.
Annotation of human cancers with EGFR signaling-associated
protein complexes using proximity ligation assays .
Sci Signal.
2015;; 8 : :ra4.. 48. Sekar
RB,
Periasamy
A. Fluorescence
resonance energy transfer (FRET) microscopy imaging of live cell protein
localizations . J Cell Biol.
2003; ; 160 :
:629.–633. 49. Xu
Y,
Piston
DW,
Johnson
CH. A
bioluminescence resonance energy transfer (BRET) system: application to
interacting circadian clock proteins . Proc Natl Acad
Sci USA.
1999; ; 96 :
:151.–156. 50. Couturier
C,
Deprez
B. Setting up a
bioluminescence resonance energy transfer high throughput screening assay to
search for protein/protein interaction inhibitors in mammalian
cells . Front Endocrinol.
2012; ; 3 : :100.. 51. Malovannaya
A,
Lanz
RB,
Jung
SY
et al.
Analysis of the human endogenous coregulator
complexome . Cell.
2011; ; 145 :
:787.–799. 52. Rual
JF,
Venkatesan
K,
Hao
T
et al.
Towards a proteome-scale map of the human protein-protein
interaction network . Nature.
2005; ; 437 :
:1173.–1178. 53. Stelzl
U,
Worm
U,
Lalowski
M
et al.
A human protein-protein interaction network: a resource for
annotating the proteome . Cell.
2005; ; 122 :
:957.–968. 54. Vinayagam
A,
Stelzl
U,
Foulle
R, et al.
A directed protein interaction network for investigating
intracellular signal transduction . Sci
Signal.
2011;; 4 : :rs8.. 55. Wang
J,
Huo
K,
Ma
L
et al.
Toward an understanding of the protein interaction network of the
human liver . Mol Syst Biol.
2011; ; 7 : :536.. 56. Rolland
T,
TaŞan
M,
Charloteaux
B
et al.
A proteome-scale map of the human interactome
network . Cell.
2014; ; 159 :
:1212.–1226. 57. Lambert
JP,
Tucholska
M,
Go
C
et al.
Proximity biotinylation and affinity purification are
complementary approaches for the interactome mapping of chromatin-associated
protein complexes . J Proteomics.
2015; ; 118 :
:81.–94. 58. Mackmull
MT,
Klaus
B,
Heinze
I
et al.
Landscape of nuclear transport receptor cargo
specificity . Mol Syst Biol.
2017; ; 13 : :962.. 59. Youn
JY,
Dunham
WH,
Hong
SJ
et al.
High-density proximity mapping reveals the subcellular
organization of mRNA-associated granules and bodies .
Mol Cell.
2018; ; 69 : :517.–32
e11.. 60. Jing
J,
He
L,
Sun
A
et al.
Proteomic mapping of ER-PM junctions identifies STIMATE as a
regulator of Ca2+ influx . Nat Cell
Biol.
2015; ; 17 :
:1339.–1347. 61. Mick
DU,
Rodrigues
RB,
Leib
RD
et al.
Proteomics of primary cilia by proximity
labeling . Dev Cell.
2015; ; 35 :
:497.–512. |



