| |
| Med Sci (Paris). 35(12): 975–981. doi: 10.1051/medsci/2019241.Quelles chaînes lourdes d’immunoglobulines pour quels
anticorps d’immunostimulation ? Christophe Dumet1 and Hervé Watier1,2* 1Équipe Fc receptors, antibodies and microenvironment, EA7501
GICC, Université de Tours, Tours, France 2Laboratoire d’immunologie, CHU de Tours,
Tours,
France |
Le formidable succès en cancérologie des anticorps inhibiteurs des points de contrôle de
l’immunité (immune checkpoint inhibitors) (IPCI) s’ajoute à tous les
autres succès des anticorps anticancéreux, qu’il s’agisse d’anticorps cytotoxiques
antitumoraux, d’antiprolifératifs, d’antiangiogéniques, d’antibody-drug
conjugates ou d’anticorps bispécifiques engageant les lymphocytes T. Comme
précédemment discuté [1]
(→), nous manquons de termes pour englober tout ou partie de ces
concepts thérapeutiques, le terme d’immunothérapie étant particulièrement difficile à
manier, y compris pour les IPCI. En effet, ces derniers déterminent une immunothérapie
active non spécifique, alors que les anticorps déterminent habituellement une
immunothérapie passive spécifique [1], même si ce
dernier point peut être nuancé par le fait que différents travaux utilisant des modèles
murins et différentes données cliniques ont montré l’induction d’une réponse T mémoire
par des anticorps dirigés contre la tumeur [32] (→).
(→) Voir le Forum de H. Watier, m/s n° 5, mai 2014, page
567
(→) Voir la Nouvelle de M. Pelegrin et al., m/s
n° 5, mai 2013, page 457
Nous préférons donc le terme d’anticorps d’immunostimulation pour
englober les anticorps conçus pour réveiller le système immunitaire et déclencher une
immunité antitumorale, d’autant que ces derniers ne vont pas se limiter aux IPCI. En
effet, les anticorps d’immunostimulation entrent a priori dans deux
catégories, les IPCI qui sont des antagonistes de récepteurs inhibiteurs (anti-CTLA-4
[cytotoxic T-lymphocyte-associated protein 4], anti-PD-1
[programmed cell death 1], etc.) ou de leurs ligands membranaires
(anti-PD-L1 [programmed death-ligand 1]), et les anticorps agonistes de
récepteurs d’activation (anti-CD40, anti-CD137, etc.). Les premiers sont déjà sur le
marché alors que les seconds sont encore au stade des essais cliniques. Bien faire la distinction entre toutes les catégories d’anticorps n’est pas qu’un simple
exercice conceptuel ou sémantique. Les effets pharmacologiques attendus diffèrent en
effet d’une catégorie à l’autre, non seulement du fait de la cible, mais aussi du choix
de la chaîne lourde d’IgG qui détermine les propriétés de la région Fc et celles de la
charnière (Figure 1). En
cancérologie, il est classique de choisir l’isotype γ1 et donc la sous-classe IgG1 afin
de renforcer le potentiel de destruction tumorale, par recrutement des effecteurs de
l’immunité, humoraux (complément) et cellulaires (cellules exprimant des récepteurs pour
la région Fc des IgG ou RFcγ). Les IgG2 (isotype γ2) et les IgG4 (isotype γ4), beaucoup
moins cytolytiques, sont donc habituellement écartées. Les anticorps antagonistes de
l’EGFR (epidermal growth factor receptor) dont l’un est une IgG1
(cétuximab) et l’autre une IgG2 (panitumumab) constituent un exemple assez éloquent. Les
deux sont efficaces dans le cancer colorectal sans mutation du gène
KRAS, laissant penser que l’antagonisation de l’EGFR suffit à
obtenir l’effet thérapeutique. En revanche, seul le cétuximab s’est révélé efficace dans
les cancers de la sphère ORL (oto-rhino-laryngologiques) ; dans ce cas, l’antagonisation
de l’EGFR ne suffit sans doute pas et l’action cytolytique de l’IgG1 apparaît
déterminante pour obtenir l’effet antitumoral.
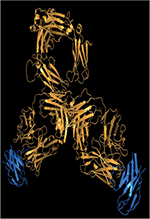
 | Figure 1. Différences entre sous-classes d’IgG humaines au niveau de la
région charnière. A et B. Structure d’une IgG1 (A) et d’une IgG4
(B), les deux seules sous-classes d’IgG humaines ayant été
cristallisées dans leur intégralité. Les chaînes
lourdes sont en noir, les chaînes légères en gris ; les N-glycanes de la
région Fc sont indiqués en rouge. Les figures ont été réalisées en utilisant
PyMOL Molecular Graphics System, version 1.7.4 (Schrödinger) à partir du
fichier PDB 1HZH représentant l’unique IgG1 humaine cristallisée, nommée B12
et dirigée contre la gp120 du VIH (virus de l’immunodéficience humaine) et à
partir du fichier PDB 5DK3 représentant le pembrolizumab, l’unique IgG4
humaine cristallisée (variant G4e1). La grande flexibilité de la région
charnière des IgG1 donne de la liberté aux bras Fab et provoque une forte
asymétrie. Il n’existe malheureusement pas de structure d’une IgG2 humaine
entière. C. Alignement de la région charnière des chaînes
lourdes g 1,
g 2 et g 4
humaines. Les acides aminés (selon la numérotation
EU) et les nucléotides qui diffèrent de la séquence des IgG1 sont indiqués
en rouge. Les cystéines engagées dans des ponts disulfures sont indiquées en
gras. Les différences notables apparaissent en fond orangé: délétion de 3
acides aminés dans l’IgG2 et l’IgG4, cystéines 219 et 220 de l’IgG2, et
sérine 228 de l’IgG4 (voir texte). |
La simple distinction entre IgG1 cytolytiques et IgG2/IgG4 non cytolytiques est en
réalité bien plus complexe. D’une part, les IgG2 et les IgG4 ne sont pas totalement
silencieuses du point de vue de l’activation du complément ou du recrutement des
cellules RFcγ+. D’autre part, les IgG1 ne se révèlent pas forcément
cytolytiques chez le patient, à l’image de quelques anticorps antagonistes tels que
l’éfalizumab (anti-intégrine LFA-1 [lymphocyte function-associated antigen
1]) ou le basiliximab (anti-IL[interleukine]-2Rα/CD25) qui ne provoquent
pas de lymphopénie. Par ailleurs, les progrès de l’ingénierie moléculaire permettent
maintenant d’obtenir des IgG1 dont l’effet cytotoxique est dopé, ou au contraire aboli
(IgG1 à Fc silencieux), ou des IgG2 et des IgG4 ayant des régions Fc totalement
silencieuses. Comme nous allons le voir, d’autres variations peuvent être introduites
pour améliorer la stabilité des anticorps in vitro (stockage
préadministration) ou in vivo. Ces variants d’ingénierie, de plus en
plus nombreux, élargissent considérablement l’éventail des possibilités
pharmacologiques. Pour les repérer plus facilement, nous avons proposé un nouveau
système de nomenclature dénommé Ge (IgG engineered), décliné en G1e,
G2e et G4e selon la sous-classe, inspiré du système de nomenclature des allotypes Gm
(IgG marker) [2]. Les Tableaux I
(variants G1e des IgG1) et II (variants G4e des IgG4) donnent une
version actualisée des variants d’anticorps ou de protéines de fusion contenant une
région Fc, au fur et à mesure de leur mise sur le marché. La diversité des cibles des anticorps d’immunostimulation jointe à la diversité des
sous-classes d’IgG et de leurs variants provoque un paysage complexe, mais qui finit par
être assez informatif sur la pharmacologie de ces anticorps chez l’homme. C’est ce que
nous allons décrire, en considérant chaque cible ou type de cible. |
Antagonistes des points de contrôle de l’immunité Anticorps anti-CTLA-4 Avec le recul, le cas des anticorps anti-CTLA-4 (CD152) est très instructif et
illustre toute la difficulté à prédire l’activité d’une sous-classe d’anticorps
chez l’homme. En 2007, deux anticorps étaient en compétition, l’ipilimumab, qui
est une IgG1κ (chaîne lourde γ1 non mutée G1e0, allotype rare G1m3,1) et le
trémélimumab qui est une IgG2κ (chaîne lourde γ2 non mutée G2e0, allotype
G2m-23). Le choix de l’IgG2 rassurait [ 3], car il correspondait mieux au profil pharmacologique
attendu d’un anticorps purement antagoniste, peu susceptible de détruire les
cellules exprimant CTLA-4. à l’inverse, le choix d’une IgG1 inquiétait, car
susceptible d’induire des effets non recherchés, à commencer par la destruction
des lymphocytes que l’on souhaitait réveiller, même si les essais précliniques
n’étaient pas source d’inquiétude [ 4]. La suite est maintenant connue… De façon a priori paradoxale,
l’ipilimumab a obtenu une AMM dans le traitement du mélanome dès 2007, tandis
que le trémélimumab n’a toujours pas été approuvé, malgré de multiples essais de
phase III dont aucun ne s’est révélé concluant. Rien ne prouve évidemment que
l’incapacité des IgG2 à recruter les effecteurs du système immunitaire soit la
cause de l’échec du trémélimumab, mais l’hypothèse mérite sérieusement d’être
soulevée. Autrement formulé, comment expliquer qu’un IPCI doublé d’une activité
déplétante soit plus actif qu’un anticorps simplement antagoniste de CTLA-4 ? Un
premier élément d’explication est venu d’expériences pratiquées chez la souris,
avec un anticorps anti-CTLA-4 décliné en plusieurs sous-classes d’IgG de souris
[5]. Dans ces
modèles, l’anticorps de sous-classe murine IgG2a (avec activité déplétante) a
montré une activité antitumorale, au contraire du même anticorps avec la
sous-classe murine IgG1 connue pour sa faible activité cytotoxique [5]. La différence d’efficacité semble
s’expliquer par la capacité de l’IgG2a anti-CTLA-4 à éliminer rapidement les
lymphocytes T régulateurs (Treg) intratumoraux, à la différence de l’IgG1 murine
anti-CTLA-4 [5]. Il faut évidemment se
garder de toute transposition hâtive avec la situation clinique (ipilimumab
vs trémélimumab), tant les sous-classes d’IgG et les
mécanismes effecteurs divergent entre espèces. De fait, comme souvent, les
données cliniques peinent à confirmer l’hypothèse. Ainsi, après des premières
observations encourageantes [6], un article récent a montré que les tumeurs de patients traités
par ipilimumab ne montraient pas d’infiltration moindre de Treg par rapport aux
tumeurs de patients traités par trémélimumab [7]. Cette étude présente néanmoins un
certain nombre de limites qui ont été discutées [8] et qui empêchent d’en tirer des
conclusions définitives. Un autre élément dont il faut tenir compte est la très
grande variabilité de réponse à l’ipilimumab, avec une faible proportion de
patients répondeurs. Or les tumeurs des patients répondeurs à l’ipilimumab
montrent avant traitement une plus forte infiltration de macrophage
CD68+ [6] et de lymphocytes
CD56+ [9]
que les non-répondeurs. Ces effecteurs cellulaires expriment le RFcγIIIa/CD16a,
un des récepteurs Fc des IgG dont nous avons révélé l’importance pour l’activité
clinique des anticorps cytotoxiques, par le biais de l’analyse du polymorphisme
de son gène et la mise en évidence d’une relation génotype/phénotype [10]. Avec le recul de
plusieurs cohortes de patients traités par différents anticorps cytotoxiques,
les patients qui répondent le mieux sont les homozygotes pour la forme
158-valine (158V) du RFcγIIIa (RFcγIIIa-158VV), qui constituent entre 15 et 20 %
de la population. Or il apparaît que, sous traitement par ipilimumab, les
patients RFcγIIIa-158VV dont la tumeur présente une forte charge néoantigénique
ont une meilleure survie que les patients porteurs de l’allotype
158-phénylalanine (158F) de ce même récepteur (à l’état hétérozygote ou
homozygote) avec une forte charge néoantigénique tumorale, et que tous les
patients ayant une faible charge néoantigénique, quel que soit l’allotype
RFcγIIIa dont ils sont porteurs [11]. Ces résultats montrent que le recrutement des
effecteurs par la région Fc de l’ipilimumab est déterminante pour l’efficacité
thérapeutique, et que l’efficacité des anticorps anti-CTLA-4 peut sans doute
être optimisée, au moins chez les patients présentant une forte charge
antigénique, afin de leur permettre de répondre même quand ils sont porteurs de
l’allotype RFcγIIIa-158F. Des données précliniques montrent d’ores et déjà que
des anticorps dont la région Fc a fait l’objet d’une ingénierie afin d’accroître
la capacité à recruter les cellules RFcγ+ ont une meilleure activité
thérapeutique [12]. Des
essais cliniques ont maintenant été lancés par Agenus, avec l’AGEN1181, une IgG1
mutée anti-CTLA-4 dont l’ingénierie protéique portant sur la chaîne lourde n’a
malheureusement pas encore été divulguée [13]. Dans tous les cas, il apparaît que les anticorps anti-CTLA-4 efficaces ne sont
pas que de simples IPCI, ce qui justifie d’autant le terme plus général
d’anticorps d’immunostimulation. S’il se confirme que leur principal mécanisme
d’action est la déplétion des lymphocytes Treg, c’est même un changement de
paradigme qui s’opère, et qui ouvre la voie au développement d’anticorps
cytotoxiques dirigés contre les lymphocytes Treg, tels que des anticorps
anti-CD25 [14] ou des
anticorps anti-OX40 (CD134) [15] qui ne sont pas des IPCI, ou des anticorps anti-TIGIT [16] qui le sont. Anticorps anti-PD-1 Contrairement aux anticorps anti-CTLA-4, les anticorps anti-PD-1 satisfont sans
doute mieux à la définition stricte d’IPCI, puisqu’il s’agit réellement
d’antagoniser un récepteur inhibiteur exprimé par des lymphocytes effecteurs de
l’immunité anticancéreuse. Les modèles murins confirment d’ailleurs qu’une IgG2a
anti-PD-1 (déplétante) ne présente pas d’activité antitumorale, au contraire
d’une IgG1 murine anti-PD-1, voire d’une IgG1 murine dont les fonctions
effectrices ont été totalement abolies [ 17]. L’emploi de souris humanisées pour les RFcγ a montré
que les anticorps anti-PD-1 de sous-classe humaine IgG4 ou IgG1 aglycosylée
(mutation N297A supprimant le site de N-glycosylation et
abolissant les fonctions effectrices) ont également une bonne activité
antitumorale. De fait, avec une belle unanimité, tous les anticorps anti-PD-1
sur le marché sont des IgG4κ (pembrolizumab, nivolumab, cémiplimab, ainsi que
toripalimab et sintilimab en Chine), et le seul anticorps anti-PD-1 d’une autre
sous-classe (pidilizumab, IgG1κ) n’a jamais dépassé la phase II, probablement du
fait d’une réactivité restreinte à certaines glycoformes de PD-1 et d’une
réactivité croisée avec DLL-1 ( delta-like canonical Notch ligand
1). Ces cinq IgG4 (pembrolizumab, nivolumab, cémiplimab, toripalimab, sintilimab) ne
sont pas pour autant des IgG4 bona fide (IgG4 G4e0), mais des
IgG4 ayant fait l’objet d’une ingénierie, à savoir l’introduction d’une mutation
ponctuelle S228P dans la région charnière (G4e1, Tableau II et
Figure 1
). Il en va de même d’autres IPCI purement antagonistes, comme le
lirilumab qui cible les KIR2DL1/-L2/-L3 des lymphocytes NK. Cette mutation
n’affecte pas les propriétés de la région Fc, mais abolit la capacité des IgG4 à
former des hémi-IgG et à se réassocier avec d’autres hémi-IgG4 pour former des
anticorps bispécifiques (Fab-arm exchange) (Figure 2) [18]. Ce phénomène a été mis en
évidence in vivo avec le natalizumab, une IgG4 non mutée (G4e0)
anti-intégrine α4 [19].
Dans la mesure où cette mutation G4e1 n’est pas protégée par un brevet [18] et qu’elle est employée de longue date
(Tableau II),
elle peut être facilement utilisée pour éviter d’éventuels inconvénients dus à
ce phénomène de Fab-arm exchange, même si aucun ne lui a été
associé jusqu’à présent. Porteuses ou non de la mutation S228P, les IgG4 gardent
la propriété de se lier au RFcγI et au RFcγIIIa-158V et ne sont donc pas dénuées
de toute propriété cytolytique [20]. Les modèles précliniques suggèrent même que l’on
gagnerait en efficacité en abolissant totalement la capacité des IgG4 anti-PD-1
à recruter des cellules RFcγ+ [20]. Le tislélizumab est d’ailleurs une IgG4 anti-PD-1 actuellement
en phase III, dont la région Fc a fait l’objet d’une ingénierie complexe
renforçant la stabilité de la molécule (double mutation S228P et R409K) et
abolissant les fonctions Fc (Fc silencieux, via une quadruple
mutation E233P, F234V, L235A et D265A).
 | Figure 2. Phénomène de Fab-arm exchange, ou de
dissociation/réassociation d’hémi-IgG4.
Chaque IgG4 a tendance à se dissocier en deux au niveau de la région
charnière et de la région Fc conduisant à la formation d’un dimère
chaîne lourde (H)/chaîne légère (L), HL. Les deux hémi-IgG qui en
résultent peuvent se réassocier avec d’autres hémi-IgG4, formant des
anticorps bispécifiques, moins avides et moins susceptibles de
former des complexes immuns. Cette propriété est naturelle chez les
IgG4, mais le plus souvent, elle n’est pas souhaitable pour un
anticorps thérapeutique. |
Tableau I.
| Numérotation G1e |
Dénominations communes internationales |
Année de première mise sur le marché |
Nature de l’ingénierie de la chaîne lourde
(Numérotation Eu) |
| G1e0 |
nombreuses |
Dès 1997 |
Aucune ingénierie |
|
| G1e1 |
abatacept, bélatacept |
2005 |
Substitutions C220S, C226S, C229S, P238S |
|
| G1e2 |
romiplostim |
2008 |
Production dans E. coli (aglycosylation) |
|
| G1e3 |
aflibercept, efmoroctocog α, eftrenonacog
α |
2011 |
Délétion des cinq premiers acides aminés de
la région charnière |
|
| G1e4 |
mogamulizumab |
2012 |
Afucosylation |
|
| G1e5 |
obinutuzumab |
2013 |
Addition d’une GlcNAc bissectrice |
|
| G1e6 |
bélimumab |
2014 |
Substitution F126L |
|
| G1e7 |
védolizumab |
2014 |
Substitutions L235A et G237A |
|
| G1e8 |
atézolizumab |
2016 |
Substitution N297A |
|
| G1e9 |
durvalumab |
2017 |
Substitutions L234F, L235E, P331S |
|
| G1e10 |
risankizumab |
2019 |
Substitutions L234A, L235A, et délétion
447K |
|
| G1e11 |
eptinézumab |
2019 |
Substitutions K213A et N297A |
Système G1e des variants d’IgG1.
|
Tableau II.
| Numérotation G4e |
Dénominations communes internationales |
Année de première mise sur le marché |
Nature de l’ingénierie de la chaîne lourde
(Numérotation Eu) |
| G4e0 |
natalizumab, ibalizumab |
2004 |
Aucune ingénierie |
|
| G4e1 |
gemtuzumab ozogamicin, pembrolizumab,
nivolumab, cémiplimab |
2000 |
Substitution S228P |
|
| G4e2 (ou G2e1) |
éculizumab |
2007 |
Hybride IgG2 (avant T260) / IgG4 (après) |
|
| G4e3 |
dulaglutide |
2014 |
Substitutions S228P, F234A, L235A
et délétion
K447 |
|
| G4e4 |
ixékizumab, dupilumab |
2016 |
Substitution S228P et délétion K447 |
|
| G4e5 |
émicizumab |
2017 |
Substitutions S228P, K196Q, F296Y, E356K,
R409K, H435R, L445P et délétions G446, K447 |
Système G4e des variants d’IgG4.
|
En attendant de connaître les résultats cliniques du tislélizumab, on peut se
demander quels inconvénients les actuels anticorps IgG4 G4e1 anti-PD-1
pourraient avoir, en particulier chez les patients homozygotes pour le
RFcγIIIa-158V, les plus sensibles aux effets cytotoxiques des anticorps. Il n’y
a malheureusement pas de données actuellement disponibles, mais l’hypothèse
selon laquelle certains patients hyperprogresseurs sous anti-PD-1 soient de ce
génotype mériterait d’être étudiée, car cette situation favoriserait une
destruction de leurs lymphocytes T antitumoraux. Anticorps anti-PD-L1 PD-L1, le principal ligand de PD-1, étant exprimé à la fois sur les cellules
tumorales et sur les cellules présentatrices d’antigènes, on peut se demander
s’il faut se contenter de l’antagoniser, ou si l’on a intérêt à doubler son
activité antagoniste d’une activité cytolytique, directement antitumorale. Les
modèles précliniques sont plutôt en faveur de la seconde hypothèse [ 17]. Traduisant cette incertitude, trois
anticorps anti-PD-L1 ayant des chaînes différentes sont sur le marché.
Curieusement, toutes sont dérivées de la chaîne γ1, sans aucune ingénierie
(G1e0) pour l’avélumab, avec une mutation N297A abolissant le site de
N-glycosylation (G1e7) pour l’atézolizumab, et avec une
triple mutation L234F/L235E/P331S (G1e8) pour le durvalumab. Les variants G1e7
et G1e8 ont des Fc silencieux, ne recrutant pas les effecteurs de l’immunité. Il
serait assez instructif de comparer l’avélumab (IgG1 à Fc compétent) à l’un ou
l’autre des deux autres anticorps (IgG1 à Fc silencieux), mais ces données ne
sont malheureusement pas disponibles. L’avélumab n’est en effet indiqué que dans
le carcinome à cellules de Merkel, où son activité cytotoxique est sans doute
essentielle [ 21], et on
ne sait pas s’il est efficace dans les cancers où les deux autres anticorps sont
indiqués. Des résultats récents suggèrent aussi qu’un anticorps anti-PD-L1
hypofucosylé (G1e4), dont la liaison de sa région Fc au RFcγIIIa est accrue, est
plus efficace in vitro pour activer les lymphocytes T
CD8 + cytotoxiques ; il est cependant difficile de prédire si cet
avantage se traduira par une plus forte efficacité clinique [ 22]. Des questionnements similaires
vont se poser pour les anticorps ciblant CD47 sur les cellules cancéreuses,
anticorps destinés à empêcher la reconnaissance de CD47 par SIRPα et à restaurer
la capacité phagocytaire des macrophages. L’anticorps anti-CD47 le plus avancé
en clinique, le magrolimab, est une IgG4 G4e1, variant dont les propriétés
cytotoxiques ont été discutées plus haut, à propos des anti-PD-1. |
Agonistes de récepteurs activateurs des lymphocytes Utiliser des anticorps agonistes de récepteurs de la famille des récepteurs du TNF-a
en cancérologie est une idée ancienne, qui a été tentée pour l’instant sans succès
vis-à-vis des récepteurs de mort, notamment les récepteurs de TRAIL
(tumor-necrosis-factor-related apoptosis-inducing ligand), DR4
(mapatumumab) et DR5 (lexatumumab, tigatuzumab, conatumumab, drozitumab). Tous
étaient des IgG1 pour potentialiser l’effet cytolytique, et certains travaux ont
montré que le coengagement des RFcγ était nécessaire à leur effet proapoptotique
[23]. Dans un but
d’immunostimulation, sont recherchés des anticorps agonistes de récepteurs
d’activation leucocytaire, évidemment dénués d’effet cytolytique sur les cellules
les exprimant. Plusieurs cibles ont été identifiées, CD40 et 4-1BB (CD137), mais
aussi OX40 (CD134), CD27, GITR (CD357, glucocorticoid-induced TNFR
family-related gene), etc. L’induction d’un signal agoniste dépend
évidemment de l’épitope reconnu sur le récepteur [24], mais aussi de la capacité d’un anticorps
par définition divalent à agréger deux molécules de récepteur, et même de la
capacité de l’anticorps à coengager des RFcg, non pas pour stimuler des fonctions
effectrices mais pour provoquer un niveau supplémentaire d’agrégation
(clustering) des récepteurs ciblés et accroître ainsi la force
du signal agoniste. Les modèles murins ont notamment mis en avant l’intérêt
d’utiliser des IgG1 murines plutôt que des IgG2a, et ont souligné l’importance du
RFcg inhibiteur, le RFcγIIb. Malgré tout, la transposition en clinique est loin
d’être simple ; aucun anticorps d’immuno-stimulation de type agoniste n’est encore
approuvé et aucun n’a d’ailleurs encore atteint la phase III. Dans tous les cas, la
plus grande prudence est de mise du fait du risque d’induction de réponses
cytokiniques massives lors des perfusions. Ce fut notamment le cas avec le TGN1412,
une IgG4 anti-CD28 superagoniste qui a déclenché un orage cytokinique chez des
volontaires sains. Cet essai désastreux a néanmoins permis de pousser plus loin les
analyses mécanistiques, révélant que l’agrégation de CD28 provoquée par le TGN1412 a
sans doute été facilitée par le coengagement du RFcγIIb humain, bien qu’il s’agisse
d’une IgG4 [25]. Anticorps anti-CD40 Le sélicrélumab (CP-870,893) fut le premier anticorps anti-CD40 agoniste testé en
clinique et a donné quelques résultats encourageants, notamment dans le cancer
du pancréas [ 26]. Cette
IgG2 ne relevait pas d’un choix délibéré d’un isotype de chaîne lourde car
l’anticorps ne faisait que dériver de souris transgéniques productrices d’IgG2
humaines ; il avait juste été sélectionné pour sa forte activité agoniste. Il
est en réalité apparu secondairement que le choix de la sous-classe IgG2
favorisait l’effet agoniste [ 27]. En effet, la région charnière de l’IgG2 est plus courte que
celle des IgG1 et comprend deux cystéines supplémentaires permettant la
formation de deux autres ponts disulfures (Figure 1), rendant l’IgG2 très rigide, surtout quand
elle adopte l’isoforme IgG2B [ 27]. Cette
rigidité accentue l’agrégation du récepteur et, en conséquence, l’effet
agoniste. Dans des modèles de souris transgéniques exprimant CD40 et les RFcγ
humains, un variant du sélicrélimab dont la région Fc a une affinité accrue pour
le RFcγIIb, mais pas pour le RFcγIIa, a montré un effet antitumoral plus
puissant encore, mais au prix d’une toxicité accrue [ 28]. En effet, l’utilisation du
sélicrélumab s’est accompagnée du syndrome de libération de cytokines, ainsi que
de toxicités plaquettaires et hépatiques. Hormis l’anticorps CDX-1140, qui est
aussi une IgG2, les autres compagnies pharmaceutiques développant des anticorps
anti-CD40 agonistes ont fait d’autres choix, notamment celui d’une IgG1 G1e0 non
mutée pour le dacétuzumab et le mitazalimab. Quant aux compagnies qui
développent des anticorps anti-CD40 antagonistes à visée immunosuppressive,
l’une a fait le choix d’une IgG4 G4e1 (blésélumab), une autre celui d’une IgG1 à
Fc silencieux et à liaison augmentée au récepteur Fc néonatal, FcRn, pour
obtenir un accroissement de demi-vie plasmatique (ravagalimab), et une autre
celui d’une IgG1 à Fc silencieux et présentant la mutation ponctuelle S131A
(iscalimab), mutation dont on ignore la raison d’être et les conséquences
pharmacologiques ! Autant dire qu’il y en a pour tous les goûts, ce qui traduit
l’incertitude quant au choix de la meilleure chaîne lourde… Anticorps anti-4-1BB (CD137) L’urélumab et l’utomilumab sont deux anticorps agonistes de la molécule
activatrice 4-1BB. Ils se distinguent à la fois par leur liaison au 4-1BB [ 29] et par leur chaîne
lourde, ce qui rend difficile leur comparaison. L’urélumab est une IgG4 G4e1
(S228P) purement agoniste (ne bloquant pas la liaison du ligand) tandis que
l’utomilumab est une IgG2 agoniste et antagoniste, bloquant la liaison du ligand
endogène mais activant néanmoins les cellules exprimant 4-1BB. Lors des essais
cliniques, l’urélumab (agoniste fort, IgG4 G4e1) s’est montré nettement plus
toxique (toxicité hépatique) que l’utomilumab (agoniste faible, IgG2), qui ne
s’est pas révélé très efficace. L’observation ne milite donc pas dans le sens
d’IgG2 qui seraient toujours plus agonistes et plus à risque de toxicité ; il y
a cependant trop de différences entre les deux produits pour en tirer une
conclusion solide. Le LVGN6051 est un nouvel anticorps anti-4-1BB conçu comme un
agoniste faible (comme l’utomilumab), mais dont la région Fc a fait l’objet
d’une ingénierie pour accroître sa liaison au RFcγIIb, sans que ni l’isotype de
chaîne lourde, ni l’ingénierie Fc ne soient divulgués [ 30]. Pour les autres cibles, comme pour
CD40 et 4-1BB, la plus grande perplexité règne sur le choix de l’isotype de
chaîne lourde, tel qu’illustré pour les anticorps anti-OX-40 [ 31]. |
De toute évidence, le rationnel qui sous-tend le choix des sous-classes d’IgG ou de
leurs variants pour les anticorps d’immunomodulation se construit au fur et à mesure
des résultats des développements cliniques. Les modèles animaux s’avèrent en effet
peu prédictifs des résultats thérapeutiques et des mécanismes d’action chez les
patients. S’il est plutôt facile de concevoir un IPCI purement antagoniste, à
l’image des anti-PD-1, la situation se complique si ces IPCI ciblent des antigènes
exprimés par les cellules tumorales (anti-PD-L1) ou des lymphocytes Treg
(anti-CTLA-4), où il peut y avoir intérêt à avoir des anticorps capables de recruter
des RFcγ. La situation est encore plus complexe pour les anticorps agonistes, pour
lesquels le choix de l’épitope, de la région charnière et de la région Fc vont
intervenir, afin de tenter d’obtenir un délicat équilibre entre efficacité et effets
indésirables. Pour ces derniers, seul l’avenir nous apprendra quel devait être le
meilleur choix ! |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les
données publiées dans cet article.
|
Ce travail est soutenu par le Ministère de l’Enseignement Supérieur et de la
Recherche, programme « Investissements d’Avenir », LabEx MAbImprove
ANR-10-LABX-53-01.
|
1. Watier
H.. Biothérapies,
immunothérapies, thérapies ciblées, biomédicaments. De quoi faut-il parler
? . Med Sci (Paris).
2014; ; 30:
:567.–575. 2. Pottier
J,
Chastang
R,
Dumet
C,
Watier
H. Rethinking the
INN system for therapeutic antibodies .
MAbs.
2017; ; 9:
:5.–11. 3. Ribas
A.. Anti-CTLA4
antibody clinical trials in melanoma . Update
Cancer Ther.
2007; ; 2:
:133.–191. 4. Keler
T,
Halk
E,
Vitale
L, et al.
Activity and safety of CTLA-4 blockade combined with vaccines in
cynomolgus macaques . J
Immunol.
2003; ; 171:
:6251.–6259. 5. Selby
MJ,
Engelhardt
JJ,
Quigley
M, et al.
Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor
activity through reduction of intratumoral regulatory T
cells . Cancer Immunol Res.
2013; ; 1:
:32.–42. 6. Romano
E,
Kusio-Kobialka
M,
Foukas
PG, et al.
Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T
cells ex vivo by nonclassical monocytes in melanoma
patients . Proc Natl Acad Sci
USA.
2015; ; 112:
:6140.–6145. 7. Sharma
A,
Subudhi
SK,
Blando
J, et al.
Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T
cells (Tregs) in human cancers . Clin Cancer
Res.
2019; ; 25:
:1233.–1238. 8. Quezada
SA,
Peggs
KS. Lost in
translation: deciphering the mechanism of action of anti-human
CTLA-4 . Clin Cancer Res.
2019; ; 25:
:1130.–1132. 9. Sanseviero
E,
O’Brien
EM,
Karras
JR, et al.
Anti-CTLA-4 activates intratumoral NK cells and combined with
IL15/IL15Rα complexes enhances tumor control .
Cancer Immunol Res.
2019; ; 7:
:1371.–1380. 10. Cartron
G,
Dacheux
L,
Salles
G, et al.
Therapeutic activity of humanized anti-CD20 monoclonal antibody
and polymorphism in IgG Fc receptor FcγRIIIa gene .
Blood.
2002; ; 99:
:754.–758. 11. Arce
Vargas
F,
Furness
AJS,
Litchfield
K, et al.
Fc effector function contributes to the activity of human
anti-CTLA-4 antibodies . Cancer
Cell.
2018;; 33:
:649.–63. 12. Ha
D,
Tanaka
A,
Kibayashi
T, et al.
Differential control of human Treg and effector T cells in tumor
immunity by Fc-engineered anti-CTLA-4 antibody .
Proc Natl Acad Sci USA.
2019; ; 116:
:609.–618. 13. Waight
J,
Manrique
M,
Gombos
R, et al.
Preclinical functional characterization of AGEN1181, a clinical
stage Fc-engineered anti-CTLA-4 antibody for the treatment of patients with
early and advanced malignancies . J Clin
Oncol.
2019; ; 37: (15 suppl)
:e14126.. 14. Arce
Vargas
F,
Furness
AJS,
Solomon
I, et al.
Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T
cells and synergizes with PD-1 blockade to eradicate established
tumors . Immunity.
2017;; 46:
:577.–86. 15. Bulliard
Y,
Jolicoeur
R,
Zhang
J, et al.
OX40 engagement depletes intratumoral Tregs via activating FcγRs,
leading to antitumor efficacy . Immunol Cell
Biol.
2014; ; 92:
:475.–480. 16. Leroy
X,
Hoofd
C,
Cuende
J, et al.
A-TIGIT antagonist antibody EOS884448 shows dual mechanism of
action by restoration of T cell effector functions and preferential
depletion of Treg . Cancer
Res.
2018;; 78 ((13) suppl):
:LB.–114. 17. Dahan
R,
Sega
E,
Engelhardt
J, et al.
FcγRs modulate the anti-tumor activity of antibodies targeting
the PD-1/PD-L1 axis . Cancer
Cell.
2015; ; 28:
:285.–295. 18. Dumet
C,
Pottier
J,
Gouilleux-Gruart
V,
Watier
H. Insights into the
IgG heavy chain engineering patent landscape as applied to IgG4 antibody
development . Mabs.
2019 ( sous presse.). 19. Labrijn
AF,
Buijsse
AO, van den
Bremer
ET, et al.
Therapeutic IgG4 antibodies engage in Fab-arm exchange with
endogenous human IgG4 in vivo . Nat
Biotechnol.
2009; ; 27:
:767.–771. 20. Zhang
T,
Song
X,
Xu
L, et al.
The binding of an anti-PD-1 antibody to FcγRI has a profound
impact on its biological functions . Cancer
Immunol Immunother.
2018; ; 67:
:1079.–1090. 21. Boyerinas
B,
Jochems
C,
Fantini
M, et al.
Antibody-dependent cellular cytotoxicity activity of a novel
anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor
cells . Cancer Immunol Res.
2015; ; 3:
:1148.–1157. 22. Goletz
C,
Lischke
T,
Harnack
U, et al.
Glyco-engineered anti-human programmed death-ligand 1 antibody
mediates stronger CD8 T cell activation than its normal glycosylated and
non-glycosylated counterparts . Front
Immunol.
2018; ; 9: :1614.. 23. Wilson
NS,
Yang
B,
Yang
A, et al.
An Fcγ receptor-dependent mechanism drives antibody-mediated
target-receptor signaling in cancer cells .
Cancer Cell.
2011; ; 19:
:101.–113. 24. Yu
X,
Chan
HTC,
Orr
CM, et al.
Complex interplay between epitope specificity and isotype
dictates the biological activity of anti-human CD40
antibodies . Cancer Cell.
2018; ; 33:
:664.–675. 25. Bartholomaeus
P,
Semmler
LY,
Bukur
T, et al.
Cell contact–dependent priming and Fc interaction with
CD32+ immune cells contribute to the TGN1412-triggered
cytokine response . J
Immunol.
2014; ; 192:
:2091.–2098. 26. Beatty
GL,
Chiorean
EG,
Fishman
MP, et al.
CD40 agonists alter tumor stroma and show efficacy against
pancreatic carcinoma in mice and humans .
Science.
2011; ; 331:
:1612.–1616. 27. White
AL,
Chan
HT,
French
RR, et al.
Conformation of the human immunoglobulin G2 hinge imparts
superagonistic properties to immunostimulatory anticancer
antibodies . Cancer Cell.
2015; ; 27:
:138.–148. 28. Dahan
R,
Barnhart
BC,
Li
F, et al.
Therapeutic activity of agonistic, human anti-CD40 monoclonal
antibodies requires selective FcγR engagement .
Cancer Cell.
2016; ; 29:
:820.–831. 29. Chin
SM,
Kimberlin
CR,
Roe-Zurz
Z, et al.
Structure of the 4–1BB/4-1BBL complex and distinct binding and
functional properties of utomilumab and urelumab .
Nat Commun.
2018; ; 9: :4679.. 30. Qi
X,
Li
F,
Wu
Y, et al.
Optimization of 4–1BB antibody for cancer immunotherapy by
balancing agonistic strength with FcγR affinity .
Nat Commun.
2019; ; 10: :2141.. 31. Zhang
D,
Goldberg
MV,
Chiu
ML. Fc Engineering
approaches to enhance the agonism and effector functions of an anti-OX40
antibody . J Biol Chem.
2016; ; 291:
:27134.–27146. 32. Pelegrin
M,
Gros
L,
Piechaczyk
M. Des effets
vaccinaux pour les anticorps monoclonaux antiviraux: une nouvelle
perspective thérapeutique ? . Med Sci
(Paris).
2013; ; 29:
:457.–460. |



