| |
| Med Sci (Paris). 35(12): 1189–1193. doi: 10.1051/medsci/2019212.Dénominations et indications des anticorps face à la
réglementation et aux pratiques des laboratoires pharmaceutiques Laura Foucault-Fruchard,1,2* Hervé Watier,1,3 and Daniel Antier1,2 1CHRU de Tours, 37000Tours,
France 2UMR 1253, iBrain, Université de Tours, Inserm,
37000Tours,
France 3EA 7501 - Groupe Innovation et ciblage cellulaire (GICC),
Université de Tours, 37000Tours,
France |
Ces dernières années, le développement des anticorps thérapeutiques a suscité un réel
engouement dans le milieu de l’industrie pharmaceutique, motivé par la croissance rapide
des ventes de ces biomédicaments. Outre la révolution apportée dans le domaine de
l’oncologie, les anticorps thérapeutiques ont prouvé leur efficacité dans la prise en
charge de maladies avec de réels besoins non satisfaits dans nombre de domaines de la
médecine. En 2019, les anticorps monoclonaux (AcM) thérapeutiques présents sur le marché français
comptent plus de 80 spécialités et 60 dénominations communes internationales (DCI)
différentes, médicaments bénéficiant d’une autorisation temporaire d’utilisation inclus.
Les dénominations et les indications thérapeutiques de ces produits répondent à la
réglementation, mais traduisent aussi les pratiques des laboratoires pharmaceutiques.
Dans cette revue, nous exposerons les cas où les stratégies commerciales développées
autour d’anticorps thérapeutiques se sont appuyées sur les noms commerciaux. |
Une dénomination commune internationale identique ; des commercialisations
différentes et des raisons diverses Historiquement, le premier cas remonte à la fin des années 1990 avec le rituximab,
premier AcM anti-CD20, commercialisé aux États-Unis sous le nom de
Rituxan® par un laboratoire américain, et sous le nom de
Mabthera® dans les autres pays du monde par une grande compagnie
pharmaceutique européenne, dont le laboratoire américain est une filiale. Deux noms
commerciaux différents, l’un enregistré par la Food and Drug
Administration (FDA) et l’autre par l’European Medicines
Agency (EMA), pour un même produit avec les mêmes indications (lymphome
non hodgkinien, leucémie lymphoïde chronique, polyarthrite rhumatoïde, granulomatose
avec polyangéite, pemphigus vulgaris) et la même concentration (10 mg/ml). Cette
situation ne s’est plus reproduite ensuite, à l’exception de quelques cas, comme
celui du tocilizumab (Actemra®/RoActemra®) et de
l’obinutuzumab (Gazyva®/Gazyvaro®) commercialisés également
par la même compagnie pharmaceutique en Europe et par sa filiale américaine aux
États-Unis. Les noms commerciaux enregistrés par l’EMA se différencient de ceux
enregistrés par la FDA par l’affixe « –ro– », laissant suggérer que cette marque de
fabrique du laboratoire permet de le distinguer de sa filiale. Ces différences de dénomination selon les territoires ont sans doute pour objectif
d’éviter une allusion trop marquée au laboratoire sur le territoire américain. Les
autorisations de mise sur le marché (AMM) sont accordées aux noms commerciaux et
limitent la circulation trans-atlantique de ces produits. Notons également le cas
d’un biosimilaire de l’adalimumab, commercialisé sous les noms
d’Amgevita® en Europe, et d’Amjevita® aux États-Unis par
un autre laboratoire américain1. Des stratégies commerciales qui ont un impact sur les finances de la
collectivité L’alemtuzumab est un AcM humanisé dirigé contre la glycoprotéine CD52 exprimée à
la surface des lymphocytes T, des lymphocytes B et des monocytes. Cette molécule
a obtenu une AMM en 2001 pour le traitement de la leucémie lymphoïde chronique à
cellules B. Dans cette indication, l’alemtuzumab était commercialisé sous le nom
de MabCampath ® (solution à diluer pour perfusion 10 mg/ml ou 30
mg/ml) par un laboratoire américain spécialisé dans les maladies rares, filiale
d’un autre grand groupe pharmaceutique européen. En septembre 2013,
l’alemtuzumab commercialisé sous le nom de Lemtrada ® (solution à
diluer pour perfusion 12 mg / 1,2 ml) a obtenu une AMM en Europe pour le
traitement de la sclérose en plaques (SEP), suite à la publication des résultats
des études CARE-MS I et II en novembre 2012 qui ont démontré l’efficacité de ce
traitement dans la SEP récurrente-rémittente [ 1]. Cette spécialité a été développée par
les suscités, en collaboration avec un grand groupe pharmaceutique allemand. À
la demande du laboratoire américain, l’AMM européenne du MabCampath ®
a été abrogée par la Commission européenne le 8 août 2012. Le retrait de l’AMM a
suscité les critiques de la revue scientifique The Lancet dans
son éditorial de novembre 2012 [ 2]. Les auteurs ont évoqué le risque d’une augmentation du coût de
l’alemtuzumab et d’une diminution de l’accessibilité à ce traitement pour de
nombreux patients atteints de SEP. Le laboratoire a justifié cet arrêt par le
souhait de se consacrer au développement de l’alemtuzumab dans le traitement de
la SEP. Cette mesure avait pour objectif d’éviter toute utilisation de
l’alemtuzumab dans le traitement de cette maladie hors des essais cliniques. Le
laboratoire s’est engagé à mettre gratuitement à disposition des hématologues
l’alemtuzumab (Campath ® 30 mg/ml, solution à diluer pour perfusion)
pour les patients qui en auraient besoin selon une procédure d’ATU (autorisation
temporaire d’utilisation) nominative. Cette stratégie commerciale n’a pas été
sans conséquences sur le plan financier en Europe. Le coût moyen du traitement
par Lemtrada ® sur 2 ans est estimé à environ 65 000 € (variable selon
les pays), soit un tarif 40 fois plus élevé que le MabCampath ® (soit
650 €/mg versus 14 €/mg) [ 3- 5]. En France, le Lemtrada® a été commercialisé seulement en 2016 sur la
base de l’avis favorable de la commission de transparence. L’attribution d’un
service médical rendu modéré dans le traitement des patients présentant une
forme très active de SEP récurrente-rémittente malgré un traitement complet et
bien conduit de 1re ou 2e ligne a empêché le laboratoire
d’obtenir l’inscription du produit sur la liste des médicaments facturés en sus
des prestations d’hospitalisation dans le cadre de la tarification à l’activité.
Pour les patients en impasse thérapeutique qui ont reçu un premier cycle de
traitement, le laboratoire s’est engagé à fournir gracieusement le deuxième
cycle. Des noms commerciaux différents pour limiter les erreurs médicales Le dénosumab est un AcM humain qui a été commercialisé sous le nom de
Prolia ® (seringue pré-remplie à 60 mg/1 ml) et de
Xgeva ® (flacon en verre de 120 mg/1,7 ml) en France,
respectivement en 2012 et 2013. Le dénosumab cible la molécule RANKL
( receptor activator of nuclear factor-k B
ligand), s’opposant à son interaction avec son récepteur RANK et
interférant avec la formation, la fonction et la survie des ostéoclastes
responsables de la résorption osseuse. Prolia ® est donc indiqué dans
le traitement de l’ostéoporose des femmes ménopausées et chez les hommes à
risque élevé de fractures, ainsi que dans le traitement de la perte osseuse. Il
est uniquement remboursé dans l’indication « traitement de l’ostéoporose
postménopausique chez les patientes à risque élevé de fracture, en deuxième
intention, en relais d’un traitement par bisphosphonates ». Xgeva ®
est quant à lui indiqué dans la prévention des complications osseuses chez les
patients souffrant de cancers solides avec métastases osseuses. Ces deux
présentations distinctes limitent le risque de confusion entre les différentes
indications et leur dosage découle des doses requises. Ces produits doivent être
administrés par voie sous-cutanée. Sur le plan de la galénique, les excipients
qui les composent et leurs teneurs sont très proches. La valeur ajoutée du
Prolia ® réside dans un dispositif prêt à l’emploi. À dose
identique, le coût du Prolia ® est environ 30 % plus élevé que celui
du Xgeva ® (3,05 €/mg versus 2,29 €/mg). Ces
spécialités suivent un circuit de distribution identique et sont disponibles en
pharmacie de ville. Un autre cas notable est celui de l’aflibercept (VEGF [vascular
endothelial growth factor]- Trap dans la littérature scientifique),
une protéine de fusion recombinante (de 115 kDa) constituée d’ectodomaines des
récepteurs du VEGF et d’une région Fc d’immunoglobuline G1 (IgG1). Il agit comme
un récepteur leurre soluble qui se lie au VEGF-A, ainsi qu’aux ligands
apparentés, PlGF (placental growth factor) et VEGF-B. En
agissant comme piège à ligand, l’aflibercept empêche la liaison des ligands
endogènes à leurs récepteurs apparentés et, par conséquent, bloque la
signalisation induite en aval. Il présente un profil de neutralisation plus
large que d’autres anti-VEGF, comme le bévacizumab (Avastin®) et le
ranibizumab (Lucentis®). Au début des années 2000, le laboratoire
américain qui le produit a signé un accord avec un grand groupe pharmaceutique
européen pour le développement de l’aflibercept dans le domaine de la
cancérologie. Dès 2004, des essais cliniques avec cette molécule dans le
traitement de la DMLA (dégénérescence maculaire liée à l’âge) ont été initiés
par le laboratoire américain, puis, en 2006, ce laboratoire a signé un accord
avec une seconde grande compagnie pharmaceutique européenne, accord visant à
développer les indications oculaires pour le même produit. En France,
l’aflibercept est commercialisé sous les noms de Zaltrap® (solution à
diluer pour perfusion 25 mg/ml) co-développé par le laboratoire américain et le
premier grand groupe européen et d’Eylea® (solution injectable en
flacon 4 mg/100 μl) co-développé par la compagnie américaine et ce second grand
groupe. Zaltrap® dispose d’une AMM depuis 2013 dans la prise en
charge thérapeutique du cancer colorectal tandis qu’Eylea® dispose
d’une AMM depuis 2012 pour le traitement de la DMLA et de la baisse d’acuité
visuelle. Fait notable, les américains ont introduit pour le Zaltrap® une DCI
différente, ziv-aflibercept, qui n’est pas valable en Europe, afin de distinguer
le Zaltrap® de l’Eylea®, le « ziv » signifiant
probablement « Zaltrap® intravascular ». Cette
dénomination ne respecte pas le système de DCI défini par l’Organisation
mondiale de la santé (OMS). De plus, la différence de formulation entre deux
spécialités contenant une molécule identique ne peut justifier l’introduction
d’une nouvelle DCI. En effet, bien que la présentation de Zaltrap®
contienne la même molécule d’aflibercept qu’Eylea®, elle se
caractérise par une osmolarité beaucoup plus élevée (1 000 mOsm/kg
versus 300 mOsm/kg). Or, il est connu que les injections
intravitréennes de solutions hyperosmolaires (osmolarité > 500 mOsm/kg)
endommagent de manière irréversible l’épithélium pigmentaire rétinien chez le
lapin et le primate [6].
De fait, le résumé des caractéristiques du produit (RCP) de Zaltrap®
mentionne que ce produit n’est pas formulé pour être compatible avec
l’environnement intra-oculaire et que l’utilisation intravitréenne est
contre-indiquée. Cependant, les préoccupations initiales concernant la
cytotoxicité et l’innocuité à long terme des injections du Zaltrap®
en intravitréen ont été en grande partie dissipées après qu’une série d’études
n’aient pas permis d’identifier des effets indésirables oculaires et systémiques
chez l’animal puis chez l’homme [7]. Plusieurs cas publiés ont en effet démontré que le
Zaltrap® en intravitréen était sans danger et efficace pour le
traitement de diverses maladies vasculaires choriorétiniennes à court et à long
terme à la dose de 1,25 mg [8-10] et à
court terme à la dose de 2 mg [11]. Des études prospectives randomisées sur le long terme et sur
des cohortes plus importantes restent cependant nécessaires pour confirmer ces
résultats. La bonne tolérance in vivo peut s’expliquer par
l’augmentation minime de l’osmolarité du vitré après injection de
Zaltrap®. Le petit volume (0,05 à 0,08 ml) de Zaltrap®
injecté dans le corps vitré adulte (4 ml) induirait une faible hausse de
l’osmolarité intravitréenne, passant de 300 Osm/kg à environ 312 mOsm/kg. Les
modalités de préparation des seringues à partir de la solution de
Zaltrap® ont été décrites à plusieurs reprises dans la
littérature [12-14]. Le coût par injection d’Eylea® est de 675,9 €. En s’appuyant sur les
données retrouvées dans la littérature (préparation de 40 seringues de 2,5 mg à
partir d’un flacon de Zaltrap® 100 mg/4 ml), le coût par injection de
Zaltrap® s’abaisse à 7,30 €, coût du consommable et du personnel
non pris en compte. Le traitement de la DMLA par les anti-VEGF permet de stabiliser et d’améliorer la
vision des patients. Sachant que la plupart de ceux-ci ont besoin de plusieurs
injections pour traiter efficacement cette maladie, l’utilisation du
Zaltrap® est une option économique particulièrement attrayante en
comparaison des tarifs des autres anti-VEGF (Avastin®,
Lucentis®, Eylea®). Le Zaltrap® serait donc
susceptible de fournir aux cliniciens un médicament supplémentaire d’un bon
rapport coût-efficacité, notamment dans les pays à revenu faible et
intermédiaire. |
Dénominations communes internationales non identiques et ententes
anti-concurrentielles entre laboratoires Comme mentionné précédemment, la prise en charge de la DMLA peut reposer sur des
injections de Lucentis® (ranibizumab) ou d’Avastin®
(bévacizumab). Bien que le cas de ces spécialités se distingue des situations que
nous venons d’exposer puisque leurs DCI diffèrent, le Lucentis® et
l’Avastin® présentent des parentés évidentes: l’un et l’autre
dérivent de l’humanisation du même anticorps monoclonal murin dirigé contre le
VEGF-A [15]. Le paratope du
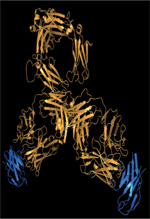
ranibizumab diffère cependant de celui du bévacizumab (Figure 1), expliquant la différence d’affinité pour
les ligands de ces deux produits [15] et,
alors que le ranibizumab est un fragment Fab, donc monovalent, le bévacizumab est
une IgG1 entière, donc bivalente.
 | Figure 1. Interaction entre VEGF et
bévacizumab/ranibizumab. Le VEGF (vascular
endothelial growth factor) est représenté en vert, le
domaine variable de la chaîne lourde de l’anticorps en bleu foncé et le
domaine variable de la chaîne légère en gris. Les 4 acides aminés du VH
qui diffèrent entre bévacizumab (ASP-29, ASN-36, HIS-109, SER-112.2) et
ranibizumab (ASP-29, HIS-36, TYR-109 et THR-112.2), numérotés selon la
nomenclature IMGT (international ImMunoGeneTics information
system), sont figurés en rouge. L’acide aminé du Vκ qui
diffère entre le bévacizumab (MET-4) et le ranibizumab (LEU-4) est
figuré en orange. Il n’est pas situé dans le paratope (d’après la
structure 6bft de la PDB, protein data bank). |
Le Lucentis® (seringue préremplie à 10 mg/ml) bénéficie d’un service
médical rendu (SMR) important dans la DMLA depuis 2007. L’Avastin®
(solution à diluer pour perfusion 25 mg/ml) est, quant à lui, autorisé dans le
traitement de plusieurs types de cancers depuis 2004. Il a été utilisé hors AMM dans
la DMLA, initialement dans un contexte où le Lucentis® n’était pas encore
disponible, puis de diminution des dépenses publiques en raison de son prix
nettement inférieur. Des études réalisées en Europe et aux États-Unis (CATT, IVAN,
MANTA, GEFAL, LUCAS et BRAMD) ont montré que le bévacizumab n’était pas inférieur au
ranibizumab en termes d’efficacité pour l’amélioration de la vision [16-21]. Le rapport bénéfice/risque ayant été jugé favorable, l’Agence nationale de sécurité
du médicament et des produits de santé (ANSM) a délivré une RTU (recommandation
temporaire d’utilisation) à l’Avastin® dans la DMLA en 2015, soit trois
ans après l’interdiction de ses injections intra-vitréennes par les pouvoirs
publics2. Un avis émis par la commission de
la transparence de la Haute autorité de santé (HAS) a statué en 2017 sur la place
identique du Lucentis® et de l’Avastin® en première intention
dans la prise en charge thérapeutique de la DMLA exsudative. Actuellement, seul le
Lucentis® est disponible en seringues pré-remplies, ce qui limite le
risque d’endophtalmie infectieuse. L’Avastin® nécessite un
reconditionnement et la préparation de seringues prêtes à l’emploi par les
pharmacies des établissements de santé autorisées à réaliser des préparations
hospitalières injectables. Ces contraintes expliquent la faible proportion des
injections d’Avastin® à ce jour par rapport au Lucentis® dans
la DMLA [22]. Face à ces décisions, la grande compagnie pharmaceutique qui produit et commercialise
l’Avastin® a ouvert des procédures de contestation en France visant à
empêcher l’utilisation de ce médicament dans la DMLA, mettant en avant le fait que
l’efficacité et la tolérance de leur produit n’avaient jamais été étudiées en
ophtalmologie. Cette requête a été rejetée par le Conseil d’État (ordonnance du 21
septembre 2015). Notons que le Lucentis® a été développé par le
laboratoire américain filiale de cette compagnie, dont les droits ont été octroyés
en Europe à une autre grande compagnie pharmaceutique européenne qui détient une
participation dans le capital de la première ! Les dispositions prises par l’Italie
en 2014 (amende d’un montant de 183 M€ aux deux compagnies) ont encouragé la Cour de
Justice de l’Union européenne (CJUE) à statuer le 23 janvier 2018 sur les pratiques
anticoncurrentielles entre ces deux grandes compagnies (mise en place d’une «
différence artificielle » entre deux médicaments ayant des propriétés thérapeutiques
équivalentes). Le 31 août 2018, la RTU établie pour l’Avastin® a été
renouvelée pour 3 ans. Le Lucentis® représente une part importante des
dépenses de l’Assurance maladie en France (en moyenne, plus de 300 millions d’euros
par an), malgré des baisses de prix successives (– 30 % entre 2014 et 2019). En
2016, le prix de la seringue de 0,10 ml d’Avastin® préparée conformément
à la RTU est passé de 10 à 100 €3. Malgré cette
augmentation, les injections de ce produit restent nettement moins onéreuses que
celles du Lucentis® (627 € par seringue). En 2018, la Cour des Comptes a
encouragé la diffusion de l’Avastin® aux patients pris en charge en ville
(économies annuelles envisagées de 500 M€) [23]. |
Ainsi, les stratégies commerciales développées autour des AcM thérapeutiques et
appuyées sur les noms commerciaux (« nom de fantaisie »)4 sont des pratiques observées dans le milieu de l’industrie
pharmaceutique. Dans les cas les plus prononcés, elles se confrontent à la
réglementation européenne et représentent un coût considérable pour la collectivité,
au regard du nombre de patients traités par les molécules concernées. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les
données publiées dans cet article.
|
Footnotes |
1. Cohen
JA,
Coles
AJ,
Arnold
DL, et al.
Alemtuzumab versus interferon beta 1a as first-line treatment for
patients with relapsing-remitting multiple sclerosis: a randomised
controlled phase 3 trial .
Lancet.
2012; ; 380:
:1819.–1828. 2. Editorial. Alemtuzumab for
multiple sclerosis . Lancet.
2012;; 380: :1792.. 3. Montgomery
SM,
Kusel
J,
Nicholas
R, et al.
Costs and effectiveness of fingolimod versus alemtuzumab in the
treatment of highly active relapsing-remitting multiple sclerosis in the UK:
re-treatment, discount, and disutility . J
Med Econ.
2017; ; 20:
:962.–973. 4. Piena
MA,
Heisen
M,
Wormhoudt
LW, et al.
Cost-minimization analysis of alemtuzumab compared to fingolimod
and natalizumab for the treatment of active relapsing-remitting multiple
sclerosis in the Netherlands . J Med
Econ.
2018; ; 21:
:968.–976. 5. Stanisic
S,
Bertolotto
A,
Berto
P, et al.
The cost-effectiveness of alemtuzumab in the management of
relapse-remitting multiple sclerosis in Italy .
GRHTA.
2019; ; 21: :S338.. 6. Marmor
MF,
Martin
LJ,
Tharpe
S, et al.
Osmotically induced retinal detachment in the rabbit and primate.
Electron miscoscopy of the pigment epithelium .
Invest Ophthalmol Vis Sci.
1980; ; 19:
:1016.–1029. 7. de Oliveira
Dias
JR,
Badaró
E,
Novais
EA, et al.
Preclinical investigations of intravitreal
ziv-aflibercept . Ophthalmic Surg Lasers
Imaging Retina.
2014; ; 45:
:577.–584. 8. Mansour
AM,
Ashraf
M,
Charbaji
A, et al.
Two-year outcomes of intravitreal
ziv-aflibercept . Br J
Ophthalmol.
2018; ; 102:
:1387.–1390. 9. Mansour
AM,
Chhablani
J,
Antonios
RS, et al.
Three-month outcome of ziv-aflibercept for exudative age-related
macular degeneration . Br J
Ophthalmol.
2016; ; 100:
:1629.–1633. 10. Chan
EW,
Eldeeb
M,
Govindhari
V, et al.
Treatment outcomes of ziv-aflibercept for treatment-naïve
polypoidal choroidal vasculopathy . Acta
Ophthalmol.
2018; ; 96:
:e258.–e259. 11. Chhablani
J,
Dedhia
CJ,
Peguda
HK, et al.
Short-term safety of 2 mg intravitreal
ziv-aflibercept . Retina.
2017; ; 37:
:1859.–1865. 12. Mansour
AM,
Al-Ghadban
SI,
Yunis
MH, et al.
Ziv-aflibercept in macular disease .
Br J Ophthalmol.
2015; ; 99:
:1055.–1059. 13. Andrade
GC,
Dias
JR,
Maia
A, et al.
Intravitreal injections of ziv-aflibercept for diabetic macular
edema: a pilot study .
Retina.
2016; ; 36:
:1640.–1645. 14. Singh
SR,
Dogra
A,
Stewart
M. Intravitreal
ziv-aflibercept: clinical effects and economic impact .
Asia Pac J Ophthalmol (Phila).
2017; ; 6:
:561.–568. 15. Magdelaine-Beuzelin
C,
Pinault
C,
Paintaud
G, et al.
Therapeutic antibodies in ophthalmology: old is new
again . MAbs.
2010; ; 2:
:176.–180. 16. CATT Research Group. ,
Martin
DF,
Maguire
MG, Ranibizumab and
Bevacizumab for neovascular age-related macular
degeneration . N Engl J Med.
2011; ; 364:
:1897.–1908. 17. Chakravarthy
U,
Harding
SP,
Rogers
CA, et al.
IVAN 2-years Alternative treatments to inhibit VEGF in
age-related choroidal neovascularisation: 2-year findings of the IVAN
randomised controlled trial .
Lancet.
2013; ; 2012: (382)
:1258.–1267. 18. Krebs
I,
Schmetterer
L,
Boltz
A, et al.
A randomised double-masked trial comparing the visual outcome
after treatment with ranibizumab or bevacizumab in patients with neovascular
age-related macular degeneration . Br J
Ophthalmol.
2013; ; 97:
:266.–271. 19. Kodjikian
L,
Souied
EH,
Mimoun
G, et al.
Ranibizumab versus Bevacizumab for neovascular age-related
macular degeneration: results from the GEFAL non-inferiority randomized
trial . Ophtalmol.
2013; ; 120:
:2300.–2309. 20. Berg
K,
Pedersen
TR,
Sandvik
L, et al.
Comparison of ranibizumab and bevacizumab for neovascular
age-related macular degeneration according to LUCAS treat-and-extend
protocol . Ophthalmology.
2015; ; 122:
:146.–152. 21. Schauwvlieghe
AM,
Dijkman
G,
Hooymans
JM, et al.
Comparing the effectiveness of bevacizumab to ranibizumab in
patients with exudative age-related macular degeneration .
The BRAMD study. PLoS One.
2016; ; 11:
:e0153052.. 22. Chast
F.. Coût du
traitement de la DMLA: au cœur de la polémique .
Rev Prat (médecine générale).
2017; ; 975:
:121.–122. 23. Rapport annuel de la cour
des comptes . Paris: ,
2018: p. :268.. |


