 |
| |||
Med Sci (Paris). 35: 43–44. doi: 10.1051/medsci/2019183.La myopathie liée à PYROXD1:
Caractérisation clinique, histologique, et génétique 16es JSFM : Prix communication orale
2018 1IGBMC, Inserm U1258, CNRS UMR7104, Illkirch, France 2Unité de Morphologie Neuromusculaire, Institut de Myologie, Inserm
UMRS974, GH Pitié-Salpêtrière, Paris, France Corresponding author. | ||||
| ||||
Au total, 12 familles avec mutation dans le gène PYROXD1 sont décrites à ce jour [1-4]. La majorité des patients présente des symptômes de la maladie à la naissance ou dans l’enfance tandis que d’autres montrent les premiers signes cliniques à l’âge adulte. La faiblesse musculaire est progressive et concerne principalement les muscles axiaux, les muscles proximaux des membres supérieurs et inférieurs, et les muscles faciaux. Une voix nasonnée, absente chez les patients adultes, est notée dans la plupart des cas précoces. Une scoliose importante est diagnostiquée chez tous les cas néonataux. Environ la moitié des patients, dont tous les cas adultes, présente une atteinte respiratoire avec réduction de la capacité vitale. D’autres signes cliniques comprennent essentiellement un palais ogival, un ptosis, une hyperlaxité, des rétractions, et une colonne raide. | ||||
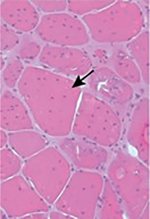
Les biopsies des patients PYRODX1 révèlent des anomalies histopathologiques similaires dont une variabilité de taille des fibres et une fibrose. La présence de groupes de fibres musculaires avec de nombreux noyaux internalisés et des lésions de type ‘core’ est retrouvée chez tous les patients et est évocatrice de la maladie [2-4] (Figure 1). Les analyses en microscopie électronique et en immunofluorescence révèlent la présence de bâtonnets et une importante désorganisation des fibres musculaires avec accumulation de protéines myofibrillaires dont la desmine, la myotiline et l’alpha-B cristalline [3-4].
| ||||
Les mutations du gène PYROXD1 ont été identifiées par séquençage d’exome, panel de gènes, et/ou Sanger. Il s’agissait de mutations faux-sens, d’épissage, et une insertion exonique de quatre nucléotides. La mutation la plus fréquente, un faux-sens c.464A>G (p.Asn155Ser), est retrouvée dans dix familles, soit à l’état homozygote, soit à l’état hétérozygote avec une deuxième mutation en trans [1-4]. De manière générale, les porteurs de mutations faux-sens homozygotes ou hétérozygotes composites (p.Asn155Ser ; p.Tyr354Cys) présentent un déficit touchant principalement les muscles des ceintures débutant dans l’enfance ou à l’âge adulte. En revanche, les patients porteurs d’une mutation d’épissage (c.285+1G>A ; c.414+1G > A ; c.415-976A > G) ont des symptômes plus sévères et plus complexes avec scoliose, voix nasonnée, hyperlaxité, rétractions et colonne raide apparaissant dès la naissance ou l’enfance. Il a été démontré que les mutations d’épissage de PYROXD1 perturbent la production d’ARN ou de protéines stables [3-4]. L’analyse des données cliniques et génétiques des 12 patients étudiés montre que les mutations conduisant à une diminution du niveau de PYROXD1 entraînent un phénotype précoce et plus sévère. | ||||
Chez la levure, l’absence de l’orthologue de PYROXD1 augmente la sensibilité au stress oxydatif [3]. Ce dernier est un facteur favorisant la formation de cores dans les fibres musculaires et peut restreindre/impacter la respiration mitochondriale [1,5]. Ces résultats suggèrent que l’altération de la fonction et de la structure musculaire des patients PYROXD1 est une conséquence directe d’un stress oxydatif prononcé. L’augmentation des marqueurs de stress HSP70 et glutathione réductase dans le muscle des patients est d’ailleurs en faveur de cette hypothèse [4]. | ||||
La myopathie liée à PYROXD1 est une maladie neuromusculaire cliniquement hétérogène dont l’âge de début est étroitement lié au type de mutation. Les quelques expériences fonctionnelles suggèrent que l’augmentation du stress oxydatif contribue très probablement au phénotype musculaire. Les biopsies des patients se distinguent par des groupements de fibres avec cores et plusieurs noyaux internalisés, ainsi que des accumulations de protéines myofibrillaires. Cette signature histologique particulière servira à orienter le diagnostic moléculaire des personnes atteintes de myopathie. | ||||
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. | ||||
1.
Saha
M
,
Reddy
HM
,
Salih
MA
, et al.
Impact of PYROXD1 deficiency on cellular respiration and
correlations with genetic analyses of limb-girdle muscular dystrophy in
Saudi Arabia and Sudan . Physiol Genomics.
2018;; 50:
:929.–39. 2.
Sainio
MT
,
Valipakka
S
,
Rinaldi
B
, et al.
Recessive PYROXD1 mutations cause adult-onset limb-girdle-type
muscular dystrophy . J Neurol.
2019;; 266:
:353.–60. 3.
O’Grady
GL
,
Best
HA
,
Sztal
TE
, et al.
Variants in the oxidoreductase PYROXD1 cause early-onset myopathy
with internalized nuclei and myofibrillar disorganization .
Am J Hum Genet.
2016;; 99:
:1086.–105. | ||||

