2008
→ Aller vers ANALYSE→ Aller vers SYNTHESE
Génétique des pathologies cardiaques familiales monogéniques
Si la cardiologie a été initialement plus lente que d’autres disciplines médicales à intégrer les outils de la génétique, des progrès considérables ont été réalisés au cours des 15 dernières années.
L’importance de la transmission héréditaire est maintenant clairement établie non seulement pour les maladies intéressant de façon exclusive le tissu cardiaque comme les arythmies familiales et les cardiomyopathies, mais aussi pour d’autres maladies génétiques comme certaines myopathies ou maladies métaboliques, pour lesquelles on sait aujourd’hui que, chez une petite proportion de patients, seul le cœur sera cliniquement atteint, comme pour certains porteurs de mutations de la dystrophine ou de la lamine A/C.
Dans le groupe des arythmies non associées à une anomalie structurale du tissu cardiaque, les arythmies ventriculaires forment le groupe de pathologies le mieux compris bien qu’hétérogène. Il comprend les syndromes du QT long (SQTL) et les tachycardies ventriculaires catécholergiques (TVC), qui sont des pathologies de l’enfant et de l’adulte, et le syndrome de Brugada dont les symptômes ne se développent qu’à l’âge adulte. Ces arythmies sont dues à un dysfonctionnement des canaux ioniques cardiaques et sont associées ou non à des anomalies de la conduction auriculo-ventriculaire. Les fibrillations atriales sont une pathologie fréquente de l’adulte, mais l’importance et l’identification des facteurs génétiques en cause restent encore à déterminer.
Parmi les cardiomyopathies caractérisées par une atteinte de la structure du myocarde, on distingue les cardiomyopathies hypertrophiques (CMH), les cardiomyopathies dilatées (CMD) et les dysplasies ventriculaires droites arythmogènes (DVDA). Ces pathologies se développent progressivement et les symptômes n’apparaissent généralement qu’à l’âge adulte.
Ces diverses pathologies d’origine génétique représentent les causes les plus fréquentes de mort subite cardiaque après les syndromes coronariens. Parmi les sujets décédés subitement avant l’âge de 35 ans, 74 % ne présentent pas d’anomalie de structure à l’autopsie et une arythmie peut être à l’origine du décès. En revanche, chez les sujets décédés de plus de 35 ans, 50 % présentent un syndrome coronarien et 25 % une cardiomyopathie (Chugh et coll., 2004

).
Arythmies cardiaques familiales
Depuis 1990, les connaissances apportées par la génétique moléculaire ont permis une meilleure prise en charge des patients à risque et le domaine des arythmies cardiaques a été l’un des premiers à en bénéficier. En effet, aujourd’hui la plupart des patients atteints de SQTL ou de TVC peuvent être traités efficacement par les bêta-bloquants permettant ainsi la prévention des tachycardies ventriculaires à l’origine des syncopes et des morts subites. L’identification de la mutation causale chez 70 à 80 % des patients atteints de SQTL permet la confirmation du diagnostic clinique, la détermination du statut des apparentés et le suivi des sujets atteints.
Syndromes du QT long congénital
Le syndrome du QT long (SQTL) congénital est caractérisé par un espace QT anormalement long à l’électrocardiogramme (ECG) – témoin d’une anomalie de la repolarisation cardiaque – et fréquemment accompagné de modifications de la morphologie de l’onde T. La découverte de la maladie concerne souvent des sujets jeunes qui font, dans certaines conditions (exercice physique, stress, émotion, prise de médicament), des syncopes par trouble du rythme ventriculaire, torsades de pointes et fibrillation ventriculaire, pouvant conduire à la mort subite. Si la maladie est diagnostiquée après la première syncope, et lorsque celle-ci n’est pas fatale, le traitement par les bêta-bloquants est efficace dans plus de 90 % des cas. En l’absence de traitement, la mortalité chez les patients présentant des symptômes est très forte, de l’ordre de 75 % à 10 ans (Moss et coll., 1991
). La gravité potentielle du pronostic justifie un dépistage des sujets atteints parmi les apparentés, par un ECG ou un enregistrement holter.
Le diagnostic repose sur la mesure de l’intervalle QT sur l’ECG, défini par le début de l’onde Q et la fin de l’onde T. De nombreux facteurs sont susceptibles de modifier cet intervalle dont le sexe, l’âge et surtout la fréquence cardiaque pour laquelle une correction est apportée par la formule de Bazett
1
Dans la formule de Bazett, le QTc et RR représentent respectivement le QT corrigé et la distance entre deux pics du tracé ECG habituel ; le QTc est exprimé en secondes.
[QTc = QT/RR (s)]. En l’absence de symptômes, un QTc > 0,44 s associé à une bradycardie ou une morphologie anormale de l’onde T est considéré comme diagnostique. Néanmoins, les troubles hydroélectriques comme l’hypokaliémie, certaines pathologies et la prise de certains médicaments allongent l’intervalle QT et doivent être recherchés.
Le SQTL congénital existe sous deux formes : l’une, très rare, sévère et associée à une surdité, est à transmission autosomique récessive et appelée syndrome de Jervell et Lange-Nielsen (Jervell et Lange-Nielsen, 1957
) ; l’autre, à transmission autosomique dominante et appelée syndrome de Romano et Ward, représente à elle seule plus de 95 % des cas (Romano et coll., 1963
; Ward, 1964
).
Sur les 8 gènes morbides qui ont été identifiés (LQT1-LQT8) (tableau I), 5 correspondent à des SQTL typiques et codent des sous-unités de canaux ioniques : KCNQ1 et KCNH2 codant des canaux potassiques, SCN5A codant un canal sodique, KCNE1 codant la protéine IsK (ou minK) régulatrice du canal potassique KvLQT1 codé par KCNE1, et KCNE2 codant la protéine MiRP1 (pour
minK-related peptide 1) (Splawski et coll., 2000
) (tableau I
). Plus de 450 mutations différentes sont répertoriées sur le site Internet
2
Site internet : pc4.fsm.it:81/cardmoc.
de la Société européenne de cardiologie dont 170 dans KCNQ1, 200 dans KCNH2 et plus de 50 dans SCN5A. La plupart de ces mutations sont des mutations faux-sens
3
Une mutation faux-sens entraîne l’incorporation d’un mauvais acide aminé dans la chaîne peptidique.
.
KCNQ1 et KCNH2 sont de loin les plus fréquemment mutés et une mutation de l’un de ces deux gènes est retrouvée chez plus de 60 % des patients. Les mutations de type faux-sens de ces gènes induisent une perte de fonction et une réduction de l’efflux potassique contribuant à la repolarisation cellulaire. Quant aux mutations non-sens
4
Une mutation non-sens entraîne le remplacement d’un nucléotide par un autre et engendre ainsi un codon stop.
ou assimilées, elles conduisent à une haplo-insuffisance et sont généralement associées à un phénotype moins sévère que les mutations faux-sens (Guicheney, communication personnelle).
Les mutations SCN5A responsables du SQTL sont pour la grande majorité des mutations faux sens associées à un gain de fonction et à courant sodium entrant persistant dans la cellule.
Grâce aux études des relations phénotype-génotype, il est possible d’orienter le diagnostic moléculaire en première intention vers l’étude d’un gène donné à partir de critères comme les facteurs déclenchant les accidents rythmiques, l’aspect de l’onde T ou la longueur relative du segment ST, la détection d’un trouble de la conduction associé chez le très jeune enfant (Lupoglazoff et coll., 2001
et 2004
).
Des manifestations d’arythmies et de syncopes à l’occasion de la prise de certains médicaments peuvent révéler un syndrome du QT long dit acquis. Il semble en réalité qu’un nombre conséquent de ces épisodes corresponde à des formes frustes de QT long congénital, causées soit par des mutations peu pénétrantes dont la fréquence pourrait dépasser 1/1 000 (Gouas et coll., 2004
), soit par la présence cumulée de variants relativement fréquents dans la population résultant en un faible allongement de l’intervalle QTc (Aydin et coll., 2005
;; Gouas et coll., 2005
).
Une liste de médicaments contre-indiqués est donnée à tous les patients identifiés lors des enquêtes familiales.
Tableau I Gènes responsables des arythmies cardiaques familiales
|
Protéine
|
Gène
|
Locus
|
Particularités
|
|
Syndromes du QT long (SQTL)
|
|
LQT1
|
Canal potassique
(sous-unité α KvLQT1) (IKs↓)
|
KCNQ1
|
11p15.5
|
Surdité associée : syndrome
de Jervell et Lange-Nielsen (AR*)
|
|
LQT2
|
Canal potassique
(sous-unité α HERG) (IKr↓)
|
KCNH2
|
7q36
| |
|
LQT3
|
Canal sodique cardiaque
(sous-unité α) (INa↑)
|
SCN5A
|
3p21
| |
|
LQT5
|
Canal potassique (sous-unité β
associé à KvLQT1) (IKs↓)
|
KCNE1
|
21q23
|
Surdité associée : syndrome
de Jervell et Lange-Nielsen (AR*)
|
|
LQT6
|
Sous-unité α de canaux
potassiques mal définis
(IKs↓, IKr ↓?)
|
KCNE2
|
21q23
| |
|
TS1/LQT8
|
Canal calcique de type L (ICaL)
|
CACNA1C
|
1q42-43
|
Syndrome de Timothy (autisme,
syndrome polyformatif)
|
|
Syndromes du QT court
|
|
SQT1
|
Canal potassique
(sous-unité α HERG) (IKr ↑)
|
KCNH2
|
7q36
| |
|
SQT2
|
Canal potassique
(sous-unité α KvLQT1) (IKs↑)
|
KCNQ1
|
11p15.5
| |
|
SQT3
|
Canal potassique Kir2.1 (IK1↑)
|
KCNJ2
|
17q24
| |
|
Tachycardies ventriculaires catécholergiques
|
|
CPVT1
|
Récepteur de la ryanodine de type 2
|
RYR2
|
1q42-q43
| |
|
CPVT2
|
Calséquestrine 2
|
CASQ2
|
1p13-21
| |
|
AND1/LQT7
|
Canal potassique Kir2.1 (IK1↓)
|
KCNJ2
|
17q24
|
Syndrome d’Andersen (anomalies neuromusculaires squelettiques)
|
|
LQT4
|
Ankyrine 2 ou B
|
ANK2
|
4q25
|
Phénotype très variable : SQTL, fibrillations atriales, bradycardie sinusale
|
|
Dysfonction sinusale familiale
|
|
SND
|
Canal pacemaker du noeud sinusal
|
HCN4
|
15q24-25
| |
|
Syndrome de Brugada
|
|
BS
|
Canal sodique cardiaque
(sous-unité α) (INa↓)
|
SCN5A
|
3p21
|
Troubles de la conduction associés ; progressifs dans le syndrome de Lenègre
|
IKs et IKr sont des courants potassiques sortant, INa un courant sodique entrant.
* AR : maladie transmise sur le mode autosomique récessif.
Syndromes du QT court
Ce syndrome familial a été rapporté pour la première fois en 2000 (Gussak et coll., 2000
). Il est caractérisé par un intervalle QTc inférieur à 300 ms, associé à des troubles du rythme. Ce syndrome, probablement très rare, est le pendant du syndrome du QT long. Les quatre mutations identifiées dans trois gènes de canaux ioniques cardiaques, HERG, KCNQ1 et KCNJ2, sont associées à un gain de fonction, engendrant un raccourcissement du potentiel d’action (tableau I
).
Tachycardies ventriculaires catécholergiques
Les tachycardies ventriculaires catécholergiques (TVC) sont caractérisées par des arythmies ventriculaires polymorphes de déclenchement adrénergique. Elles surviennent essentiellement chez des enfants ou adolescents et sont responsables de syncopes et de morts subites en l’absence de toute anomalie morphologique cardiaque (Coumel et coll., 1978
). L’ECG de repos, enregistré en dehors des périodes de tachycardie ventriculaire, est souvent normal. La mortalité due aux TVC en l’absence de traitement est très élevée, atteignant 30 à 50 % à l’âge de 30 ans (Leenhardt et coll., 1995
). De plus, il existe une corrélation entre l’âge de survenue de la première syncope et la sévérité de la maladie, avec un pronostic très péjoratif lorsque les pertes de connaissance surviennent chez un sujet très jeune. Les bêta-bloquants réduisent de façon significative les syncopes et la mort subite, rendant rare l’implantation de défibrillateur cardiaque.
Le diagnostic de TVC est établi après une épreuve d’effort avec enregistrement d’ECG montrant des anomalies rythmiques caricaturales de cette affection : lors d’une accélération du rythme sinusal, survenue d’extrasystoles ventriculaires, d’abord monomorphes, puis bidirectionnelles et polymorphes, suivies de salves de tachycardies ventriculaires polymorphes plus ou moins soutenues.
Les bases génétiques ont été élucidées, en 2001, par la mise en évidence de mutations dans deux protéines spécifiques du tissu cardiaque jouant un rôle déterminant dans la régulation du calcium intracellulaire lors du couplage excitation-contraction des cardiomyocytes : le récepteur de la ryanodine de type 2 (RYR2), protéine-canal responsable du relargage du calcium du réticulum sarcoplasmique, et la calséquestrine de type 2 (CASQ2), qui lie le calcium dans le réticulum sarcoplasmique (tableau I
).
Des mutations faux-sens transmises sur le mode autosomique dominant ont été identifiées dans le gène RYR2 chez environ 50 % des patients (Priori et coll., 2000
; Laitinen et coll., 2001
). Ce gène est constitué de 105 exons ; les mutations sont situées dans 3 régions fonctionnelles qui peuvent être analysées en première intention. De plus, des mutations faux-sens ou non-sens récessives ont été identifiées dans le gène codant la calséquestrine de type 2 (CASQ2) chez quelques rares cas (Lahat et coll., 2001
; Postma et coll., 2002
). Une bradycardie sinusale est retrouvée chez la plupart des patients porteurs de mutations RYR2 et CASQ2. La détection d’une bradycardie chez un enfant symptomatique doit conduire à pratiquer une épreuve d’effort (Postma et coll., 2005
).
Quelques formes de TVC associées ou non à un allongement de l’intervalle QT sont dues à des mutations entraînant une perte de fonction du gène KCNJ2, codant le canal potassique Kir 2.1. Ce gène a été initialement décrit comme responsable du syndrome d’Andersen associant des anomalies neuromusculaires et squelettiques à des troubles rythmiques (Schulze-Bahr, 2005
). Ce gène a également été aussi décrit comme le gène LQT7.
Une mutation du gène ANK2, codant l’ankyrine 2, a été récemment identifiée dans une grande famille associant une dysfonction sinusale et un allongement de l’intervalle QT (locus LQT4) (Mohler et Bennett, 2005
). Néanmoins, comme l’ankyrine B est une protéine intracellulaire associée à l’échangeur Na
+/Ca
+, à la Na
+/K
+ ATPase et au récepteur à l’inositol triphosphate (InsP3R) localisé dans les tubules transverses du réticulum sarcoplasmique, il n’est pas étonnant que certaines mutations puissent ne pas être associées à un allongement de l’intervalle QT mais à un phénotype de type TVC.
La diversité et l’importance des atteintes cardiaques associées aux mutations de KCNJ2 et ANK2, peu fréquentes semble-t-il, restent à définir.
Syndrome de Brugada
En 1992, les frères Brugada ont décrit une nouvelle entité clinique associant un aspect électrocardiographique particulier à un risque élevé de syncope et de mort subite chez des patients présentant par ailleurs un cœur structurellement normal, appelée depuis syndrome de Brugada (SB) (Brugada et Brugada, 1992
). Les anomalies électrocardiographiques sont caractérisées par un sus-décalage du segment ST dans les dérivations précordiales V1 à V3 et une morphologie du complexe QRS de type bloc de branche droit. Les épisodes de syncope sont dus à des tachycardies ventriculaires polymorphes, qui lorsqu’elles dégénèrent en fibrillation ventriculaire conduisent à la mort subite. Ces arythmies surviennent le plus souvent au repos ou durant le sommeil chez les hommes de 40 à 50 ans. Il s’agit d’une maladie arythmogène se transmettant sur le mode autosomique dominant à pénétrance incomplète.
Des consensus concernant les critères électrocardiographiques permettant le diagnostic ont été publiés en 2002 et en 2005 (Wilde et coll., 2002
; Antzelevitch et coll., 2005
). Trois types distincts d’anomalies de la repolarisation peuvent être identifiés mais un seul, le type 1, est considéré comme diagnostique en l’absence de symptômes. Il est à noter que l’ECG se normalise à un moment ou un autre chez à peu près la moitié des patients (Brugada et coll., 2001
), ce qui rend difficile le diagnostic des apparentés.
Le diagnostic du SB a été jusqu’à présent posé après exclusion de toute anomalie structurale du cœur. Les examens pratiqués sont l’ECG, l’échographie cardiaque, l’imagerie par résonance magnétique, la coronarographie et la ventriculographie gauche et droite. L’administration intraveineuse d’agents anti-arythmiques de classe I est utilisée pour démasquer les manifestations ECG typiques du SB, en particulier chez les apparentés (Hong et coll., 2004
). Ces agents ont pour conséquence la majoration de l’élévation du segment ST ou son apparition si elle était absente sur l’ECG de base.
L’implantation d’un défibrillateur automatique est le seul recours pour la prévention primaire ou secondaire de l’arrêt cardiaque chez les sujets symptomatiques. Ainsi, l’un des objectifs principaux lors de la prise en charge est l’identification des sujets à haut risque de mort subite. Dans ce contexte, la valeur pronostique de l’induction de fibrillation ventriculaire par la stimulation ventriculaire programmée est encore débattue à ce jour chez les sujets asymptomatiques.
Un traitement par la quinidine ou l’hydroquinidine (Serecor®) pourrait être une thérapie efficace pour réduire le risque de survenue d’accidents cardiaques chez les patients asymptomatiques inductibles (Hermida et coll., 2004
).
Chez 1 cas sur 5 environ, ce syndrome est provoqué par des mutations sur le gène SNC5A codant la sous-unité du canal sodique cardiaque, une protéine impliquée dans le contrôle de l’excitabilité myocardique. Ces mutations faux sens ou conduisant à la perte d’un allèle sont associées à une réduction du nombre de canaux sodiques à la membrane (Viswanathan et Balser, 2004
). Néanmoins, les frontières du SB sont encore mal définies ; si la plupart des patients porteurs de mutations SCN5A présentent un ralentissement modéré de la conduction auriculo-ventriculaire, certains peuvent avoir des troubles de la conduction progressifs, encore appelés maladie de Lenègre (Schott et coll., 1999
), une cardiomyopathie dilatée (Mc Nair et coll., 2004
), ou des anomalies structurales (Frustaci et coll., 2005
), comme cela a été aussi observé chez des souris déficientes en canaux sodiques (Royer et coll., 2005
).
D’après une étude récente, parmi les patients diagnostiqués comme présentant un syndrome de Brugada sans mutation du gène SCN5A, un nombre important présenterait des anomalies histologiques non décelables à angiographie d’origine inflammatoire ou autre (Frustaci et coll., 2005
). Ceci suggère que des biopsies myocardiques pourraient aider à une meilleure compréhension et classification de ces pathologies.
Selon certaines équipes, le syndrome de Brugada serait dû à une anomalie électrique primaire du myocarde droit pouvant entraîner une dégénération des myocytes au cours de l’évolution de la maladie (Gussak et coll., 1999
) tandis que pour d’autres, celui-ci serait une forme précoce de cardiomyopathie arythmogène du ventricule droit, c’est-à-dire la manifestation d’une cardiomyopathie masquée qui deviendrait histologiquement décelable après une période de latence (Saffitz, 2005
).
L’identification de nouveaux gènes et mutations aidera à mieux comprendre les diverses pathologies liées à un risque important de fibrillation ventriculaire ; c’est un enjeu majeur pour la recherche, rendu difficile par la rareté de grandes familles et par la complexité du diagnostic.
Cardiomyopathies
Les cardiomyopathies sont le plus souvent des pathologies se développant progressivement avec l’âge et du fait de cette pénétrance partielle, leur incidence est difficile à estimer. Dans le cadre des études familiales, l’identification des sujets asymptomatiques potentiellement atteints nécessite un ensemble d’examens complémentaires le plus souvent non invasifs comme l’ECG, l’échocardiographie ou l’IRM. Ceci est aujourd’hui facilité par les études moléculaires quand une mutation a été identifiée.
La prévalence de la cardiomyopathie hypertrophique (CMH) a été étudiée de façon prospective et évaluée à 1/500 dans une population de jeunes adultes (Maron et coll., 1995a
). Pour les cardiomyopathies dilatées (CMD) et les dysplasies ventriculaires droites arythmogènes (DVDA), les chiffres rapportés dans la littérature sont de 1/2 000 et de 1 à 2/10 000 respectivement. La transmission est le plus souvent autosomique dominante mais des formes autosomiques récessives sont aussi responsables de CMD et de DVDA.
Quand ces pathologies sont diagnostiquées chez un sujet jeune asymptomatique, le sport doit être déconseillé car il est associé à un nombre important de morts subites (Maron et coll., 1995b
; Heidbuchel et coll., 2003
).
Cardiomyopathies hypertrophiques
Les CMH sont caractérisées par une hypertrophie du ventricule gauche sans dilatation, en l’absence de toute maladie qui pourrait provoquer une augmentation de l’épaisseur de la paroi du ventricule gauche. Elle peut revêtir différents aspects cliniques et anatomiques et la sévérité est très variable, des formes asymptomatiques jusqu’aux patients présentant une insuffisance cardiaque due à une évolution vers une cardiomyopathie restrictive.
Les examens échocardiographiques en mode TM et bidimensionnel sont les examens clés permettant de reconnaître l’hypertrophie et d’en déterminer le siège. Elle touche le plus souvent le septum interventriculaire, mais parfois la pointe ou seulement la paroi latérale.
La CMH est associée à un très large spectre clinique : syncopes, vertiges, douleurs thoraciques, dyspnées, arythmies. Sa gravité est liée à la mort subite à l’effort, évaluée à 5 % (1 à 2 %) par an, et à l’accroissement de la rigidité ventriculaire qui entraîne une insuffisance cardiaque.
D’un point de vue histologique, la CMH est caractérisée par une désorganisation myocytaire, les cellules étant hypertrophiées et perdant leur orientation régulière jusqu’à former des angles droits. De nombreux sujets ont un ECG anormal parfois associé à un examen échocardiographique normal, qui peut être le reflet de la désorganisation myofibrillaire. Aucun traitement médicamenteux (bêta-bloquants, antagonistes calciques) ne s’est avéré efficace dans la régression de l’hypertrophie mais ils sont utiles pour l’amélioration des symptômes. Seul le défibrillateur implantable permet une prévention efficace de la mort subite et sera envisagée en cas de mort subite récupérée ou de syncope typique ou dans certains cas de troubles du rythme ventriculaire avec identification d’une mutation associée à un pronostic péjoratif. Dans les formes les plus sévères, une greffe cardiaque est généralement pratiquée.
La plupart des gènes impliqués ont été identifiés par génétique inverse et montrent une grande hétérogénéité allélique et génique (Seidman et Seidman, 2001
). Ces gènes codent pour des protéines du sarcomère :
• protéines de filament épais comme les chaînes lourdes et légères de la myosine ;
• protéines du filament fin comme l’actine a cardiaque, la tropomyosine et les protéines du complexe de la troponine (T, C, I) ;
• protéines jouant un rôle dans le maintien de la structure du sarcomère comme la protéine C cardiaque ou la titine (tableau II
).
Plus de 300 mutations, pour la plupart faux-sens, ont été identifiées dans le gène de la chaîne lourde bêta de la myosine, MYH7, qui fut le premier à être identifié comme responsable de la CMH (Geisterfer-Lowrance et coll., 1990
). L’autre gène majeur, MYBPC3, code la protéine C de liaison à la myosine ; les deux tiers des mutations de ce gène sont des mutations conduisant à un décalage du cadre de lecture (Bonne et coll., 1995
).
L’étude systématique de 8 gènes chez un ensemble de près de 200 patients français a montré que les gènes les plus fréquemment mutés étaient MYBPC3 (43 %), MYH7 (40 %), TNNI3 (6 %), TNNT2 (6 %) et MYL2 (4 %) (Richard et coll., 2003
).
Le profil évolutif de la maladie et sa présentation clinique peuvent être très différents d’un sujet à l’autre aussi bien pour des mutations dans des gènes différents que des mutations au sein d’un même gène voire d’une même mutation. Bien que les mutations de certains gènes semblent plus souvent associées à des accidents rythmiques, aucun schéma clair en termes de relations entre phénotype et génotype permettant d’orienter sur l’étude d’un gène donné n’a pu encore être établi (Charron et coll., 2002
).
Tableau II Gènes responsables des cardiomyopathies hypertrophiques
|
Protéine
|
Gène
|
Locus
|
Particularités
|
|
Filament épais du sarcomère
|
|
Chaîne lourde β de la myosine
|
MYH7
|
14q12
| |
|
Chaîne lourde α de la myosine
|
MYH6
|
14q12
| |
|
Chaîne légère essentielle de la myosine
|
MYL3
|
3q21.2-q21.3
| |
|
Chaîne légère régulatrice de la myosine
|
MYL2
|
12q23-q24.3
| |
|
Filament fin du sarcomère
|
|
Actine α cardiaque
|
ACTC
|
15q14
| |
|
Troponine T cardiaque
|
TNNT2
|
1q32
| |
|
Troponine I cardiaque
|
TNNI3
|
19p13.4
| |
|
Troponine C cardiaque
|
TNNC1
|
3p21.3
| |
|
α-Tropomyosine squelettique
|
TPM1
|
15q22.1
| |
|
Sarcomère et protéines associées à la strie Z
|
|
Protéine C cardiaque
|
MYBPC3
|
11p11.2
| |
|
Titine
|
TTN
|
2q35
|
Myopathie (ADa)
|
|
Protéine LIM musculaire
|
CSRP3
|
11p15.1
| |
|
Téléthonine
|
TCAP
|
17q12
|
Myopathie (ADa)
|
|
Sarcolemme
|
|
Cavéoline 3
|
CAV3
|
3p25
|
Myopathie (ADa, ARb)
|
|
Réticulum sarcoplasmique
|
|
Phospholamban
|
PLB
|
6q22.1
| |
a AD : maladie transmise sur le mode autosomique dominant ; b AR : maladie transmise sur le mode autosomique récessif
Des mutations ont été identifiées dans 13 gènes et il est pratiquement impossible à l’heure actuelle de séquencer tous ces gènes chez un même patient. Néanmoins, l’analyse des 5 gènes les plus fréquemment mutés permet de détecter la mutation causale chez 70 à 80 % des patients. De plus dans 3 à 5 % des cas, des génotypes complexes associant deux mutations hétéroalléliques ou sur deux gènes différents expliquent les phénotypes les plus sévères (Richard et coll., 2003
). Les études fonctionnelles suggèrent que les mutations des protéines sarcomériques entraînent une altération primitive de la fonction du sarcomère, avec une hypertrophie secondaire et compensatrice.
Cardiomyopathies dilatées
Les CMD sont caractérisées par une dilatation des cavités cardiaques, ventricule gauche ou ventricules gauche et droit, qui peut être considérable et par une altération de la fonction systolique gauche. L’histoire naturelle est associée à un mauvais pronostic dû à la survenue d’insuffisance cardiaque et à la possibilité de morts subites. Elles constituent une cause majeure de transplantations cardiaques et sont un problème important en santé publique tant par leur importance que par leur fréquence. Il s’agit souvent de pathologies multifactorielles et le rôle d’anomalies immunologiques, d’infections virales ou de facteurs environnementaux tels qu’une consommation excessive d’alcool est démontré depuis longtemps.
La majorité des cas sont sporadiques et auraient une origine multifactorielle associant des facteurs environnementaux et génétiques (gènes de prédisposition). Dans 20 à 35 % des cas, on détecte une forme familiale monogénique avec un mode de transmission variable (tableau III
).
Tableau III Gènes responsables des cardiomyopathies dilatées
|
Protéine
|
Gène
|
Locus
|
Particularités
|
|
Sarcomère
|
|
Chaîne lourde β de la myosine
|
MYH7
|
14q12
| |
|
Troponine T
|
TNNT2
|
1q32
| |
|
Troponine I
|
TNNI3
|
19q13.4
| |
|
Troponine C
|
TNNC1
|
3p21.1
| |
|
Actine α cardiaque
|
ACTC
|
15q14
| |
|
α-Tropomyosine squelettique
|
TPM1
|
15q22.1
| |
|
Protéine C cardiaque
|
MYBPC3
|
11p11.2
| |
|
Sarcomère et protéines associées à la strie Z
|
|
Titine
|
TTN
|
2q35
|
Myopathie
|
|
Titine-cap/ Téléthonine
|
TCAP
|
17q12
|
Myopathie
|
|
Protéine du muscle LIM
|
CSRP3
|
11p15.1
| |
|
Métavinculine
|
VCL
|
10q22.1-q23
| |
|
Protéine Zasp
|
LDB3
|
10q22.2-q23.3
|
Myopathie myofibrillaire
|
|
Cytosquelette
|
|
Dystrophine
|
DMD
|
Xp21.2
|
Myopathie
|
|
δ-Sarcoglycane
|
SGCD
|
5q33
|
Myopathie
|
|
Filaments intermédiaires
|
|
Desmine
|
DES
|
2q35
|
Myopathie
|
|
Lamine A/C
|
LMNA
|
1q21.2
|
Troubles de la conduction Myopathie
|
|
Canaux et protéines associées
| | | |
|
Canal potassique ATP dépendant
|
SUR2A/ ABCC9
|
12p12.1
|
Tachycardies ventriculaires
|
|
Phospholamban
|
PLN
|
6q22.1
| |
|
Mitochondries
|
|
Tafazzine
|
TAZ
|
Xq28
|
Syndrome de Barth
|
Il existe une grande diversité des causes génétiques incriminées (Amara et coll., 2004
). Les mutations identifiées peuvent toucher des gènes codant :
• des protéines du sarcomère et altérer la production de la force ;
• des protéines du cytosquelette et altérer la transmission de la force ;
• des protéines nucléaires et altérer notamment la stabilité nucléaire ; des mutations peuvent être également présentes sur des gènes codant d’autres protéines impliquées dans différents mécanismes intervenant dans la fonction cardiaque, que ce soit la signalisation calcique ou l’apoptose.
Ces mutations n’expliquent qu’un faible pourcentage des cas familiaux. Ce sont les mutations du gène codant la chaîne lourde de la myosine (MYH7) qui sont les plus fréquentes, mais elles sont trouvées chez moins de 10 % des cas familiaux, ce qui rend la recherche de mutation dans un contexte diagnostique très limitée, voire impossible (Chang et Potter, 2005
).
Néanmoins, dans le cas d’enquête familiale, il est important de dépister les apparentés, de diagnostiquer les formes débutantes et de rechercher les phénotypes annexes qui peuvent orienter vers un gène donné particulier comme celui de la lamine A/C en cas d’atteinte musculaire latente ou des troubles de la conduction auriculo-ventriculaire (Fatkin et coll., 1999
; Meune et coll., 2006
).
Dysplasie ventriculaire droite arythmogène
La dysplasie ventriculaire droite arythmogène (DVDA) est une cardiomyopathie du ventricule droit caractérisée par une infiltration adipeuse du myocarde avec persistance de fibres myocardiques survivantes entourées de fibrose. Elle entraîne une dilatation ventriculaire droite, localisée puis diffuse, et tardivement des manifestations d’insuffisance cardiaque. Elle peut aussi être associée à une atteinte ventriculaire gauche. Elle a pour conséquence des anomalies électriques souvent visibles sur ECG en précordiales droites : inversion de l’onde T, micro-potentiels, onde epsilon et retard de la conduction auriculo-ventriculaire qui peuvent conduire à la survenue de tachycardies ventriculaires, de syncopes et de fibrillations ventriculaires. Le risque de mort subite est élevé avant l’âge de 35 ans, en particulier lors d’exercice physique.
Le diagnostic est établi après des explorations non invasives (ECG et échocardiographie) ou invasives plus spécifiques, comme l’angioscintigraphie en contraste de phase ou l’angiographie du ventricule droit et du ventricule gauche. L’exploration électrophysiologique peut être proposée en vue d’évaluer le risque de déclenchement de tachycardies ventriculaires ou fibrillations ventriculaires. Elle permet également de cartographier les tachycardies ventriculaires en vue d’un éventuel geste d’ablation.
Les troubles du rythme ventriculaire sont le plus souvent bien contrôlés par les anti-arythmiques, les méthodes ablatives et le défibrillateur implantable.
La DVDA est transmise le plus souvent selon un mode autosomique dominant. Neuf loci ont été identifiés depuis 1994 mais c’est depuis moins de 5 ans que les premières mutations ont été identifiées dans des protéines du desmosome (Sen-Chowdhry et Syrris, 2005
) (tableau IV
). Le premier gène a été découvert grâce à la maladie de Naxos qui est une forme récessive de DVDA avec une atteinte de la peau et des cheveux : il s’agissait d’une délétion de 2 pb dans le gène de la plakoglobine (McKoy et coll., 2000
). De même, des mutations récessives ou dominantes de la desmoplakine (DSP), la protéine la plus représentée du desmosome qui fait le lien entre le desmosome et les filaments intermédiaires, ont été identifiées chez des patients (Norgett et coll., 2000
; Rampazzo et coll., 2002
; Alcalai et coll., 2003
). Mais il semble que ce soit la plakophiline 2 qui s’avère à ce jour la protéine la plus fréquemment mutée dans la DVDA classique. Dans une série de 120 propositus, une mutation a été retrouvée chez 25 % d’entre eux (Gerull et coll., 2004
).
Tableau IV Gènes responsables des dysplasies ventriculaires droites arythmogènes (DVDA)
|
Localisation
|
Protéine
|
Gène
|
Locus
|
Particularités
|
|
Desmosome
|
Plakoglobine
|
JUP
|
17q21
|
Maladie de Naxos (AR*)
|
| |
Desmoplakine
|
DSP
|
6p24
|
Syndrome de Carjaval (AR*)
|
| |
Plakophiline-2
|
PKP2
|
12p11
| |
|
Réticulum sarcoplasmique
|
Récepteur à la ryanodine de type 2
|
RYR2
|
1q42-q43
| |
|
Cytokine Rôle sur la matrice extracellulaire ou sur la stabilité des jonctions intercellulaires ?
|
Transforming growth factor β3
|
TGFB 3
|
14q23-q24
|
Mutations dans les régions régulatrices conduisant à une surexpression
|
* AR : maladie transmise sur le mode autosomique récessif
Quelques mutations ont été rapportées dans le gène codant le récepteur de la ryanodine cardiaque,
RYR2 (Tiso et coll., 2001
), et dans le gène
TGF (Beffagna et coll., 2005
).
Il y a encore peu de mutations identifiées à ce jour ; ceci ne permet pas d’établir des relations phénotype-génotype mais il est certain que les connaissances vont croître rapidement pour ces pathologies dans les prochaines années. L’identification des mutations dans les familles permettra de détecter les porteurs asymptomatiques, de les suivre et de leur contre-indiquer une activité sportive intense. De plus, on peut espérer que certaines anomalies électrocardiographiques pourront constituer des marqueurs pour orienter le diagnostic moléculaire.
En conclusion, la probabilité d’identifier une mutation pour un patient atteint d’un trouble du rythme ou d’une cardiomyopathie d’origine génétique est aujourd’hui importante pour un patient atteint du SQTL ou d’une CMH, car pour chacune de ces deux pathologies il y a deux gènes majeurs – KCNQ1 et KCNH2 pour le SQTL et MYH7 et MyBPC3 pour la CMH – qui sont mutés dans 60 à 70 % des cas. Cette probabilité est moindre pour les autres pathologies.
Néanmoins, les connaissances sur les protéines responsables de ces pathologies progressent rapidement et nous aident à mieux les définir d’un point de vue nosologique, à envisager une prise en charge plus précoce des patients et le développement de nouvelles thérapies.
Bibliographie
[1] alcalai r,
metzger s,
rosenheck s,
meiner v,
chajek-shaul t. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair.
J Am Coll Cardiol. 2003;
42:319
-327

[2] amara me,
villard e,
komajda m. Mise au point de la cardiomyopathie dilatée familiale.
Ann Cardiol Angéiol. 2004;
54:151
-156
[3] antzelevitch c,
brugada p,
borggrefe m,
brugada j,
brugada r, et coll.. Brugada syndrome: report of the second consensus conference.
Heart Rhythm. 2005;
2:429
-440
[4] aydin a,
bahring s,
dahm s,
guenther up,
uhlmann r, et coll.. Single nucleotide polymorphism map of five long-QT genes.
J Mol Med. 2005;
83:159
-165
[5] beffagna g,
occhi g,
nava a,
vitiello l,
ditadi a, et coll.. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1.
Cardiovasc Res. 2005;
65:366
-373
[6] bonne g,
carrier l,
bercovici j,
cruaud c,
richard p, et coll.. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy.
Nat Genet. 1995;
11:438
-440
[7] brugada p,
brugada j. Right bundle branch block, persistent ST elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome.
J Am Coll Cardiol. 1992;
20:1391
-1396
[8] brugada p,
brugada j,
brugada r. Dealing with biological variation in the Brugada syndrome.
Eur Heart J. 2001;
22:2231
-2232
[9] chang an,
potter jd. Sarcomeric protein mutations in dilated cardiomyopathy.
Heart Fail Rev. 2005;
10:225
-235
[10] charron p,
heron d,
gargiulo m,
richard p,
dubourg o, et coll.. Genetic testing and genetic counselling in hypertrophic cardiomyopathy: the French experience.
J Med Genet. 2002;
39:741
-746
[11] chugh ss,
jui j,
gunson k,
stecker ec,
john bt, et coll.. Current burden of sudden cardiac death: multiple source surveillance versus retrospective death certifi-cate-based review in a large U.S. community.
J Am Coll Cardiol. 2004;
44:1268
-1275
[12] coumel p,
fidelle j,
lucet v,
attuel p,
bouvrain y. Catecholamine-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases.
Brit Heart J. 1978;
40:28
-37
[13] fatkin d,
macrae c,
sasaki t,
wolff mr,
porcu m, et coll.. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease.
N Engl J Med. 1999;
341:1715
-1724
[14] frustaci a,
priori sg,
pieroni m,
chimenti c,
napolitano c, et coll.. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome.
Circulation. 2005;
112:3680
-3687
[15] geisterfer-lowrance aa,
kass s,
tanigawa g,
vosberg hp,
mckenna w, et coll.. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation.
Cell. 1990;
62:999
-1006
[16] gerull b,
heuser a,
wichter t,
paul m,
basson ct, et coll.. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy.
Nature Genet. 2004;
36:1162
-1164
[17] gouas l,
bellocq c,
berthet m,
potet f,
demolombe s, et coll.. New KCNQ1 mutations leading to haploinsufficiency in a general population; Defective trafficking of a KvLQT1 mutant.
Cardiovasc Res. 2004;
63:60
-68
[18] gouas l,
nicaud v,
berthet m,
forhan a,
tiret l, et coll.. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population.
Eur J Hum Genet. 2005;
13:1213
-1222
[19] gussak i,
antzelevitch c,
bjerregaard p,
towbin ja,
chaitman br. The Brugada syndrome: clinical, electrophysiologic and genetic aspects.
J Am Coll Cardiol. 1999;
33:5
-15
[20] gussak i,
brugada p,
brugada j,
wright rs,
kopecky sl, et coll.. Idiopathic short QT interval: a new clinical syndrome?.
Cardiology. 2000;
94:99
-102
[21] heidbuchel h,
hoogsteen j,
fagard r. High prevalence of right ventricular involvement in endurance athletes with ventricular arrhythmias. Role of an electrophysiologic study in risk stratification.
Eur Heart J. 2003;
24:1469
-1470
[22] hermida js,
denjoy i,
clerc j,
extramiana f,
jarry g, et coll.. Hydroquinidine therapy in Brugada syndrome.
J Am Coll Cardiol. 2004;
43:1853
-1860
[23] hong k,
brugada j,
oliva a,
berruezo-sanchez a,
potenza d, et coll.. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syn-drome caused by SCN5A mutations.
Circulation. 2004;
110:3023
-3027
[24] jervell a,
lange-nielsen f. Congenital deaf mutism, functional heart disease with prolongation of the QT interval and sudden death.
Am Heart J. 1957;
54:59
-68
[25] lahat h,
pras e,
olender t,
avidan n,
ben-asher e, et coll.. A missense muta-tion in a highly conserved region of CASQ2 is associated with autosomal recessive cate-cholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel.
Am J Hum Genet. 2001;
69:1378
-1384
[26] laitinen pj,
brown km,
piippo k,
swan h,
devaney jm, et coll.. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia.
Circulation. 2001;
103:485
-490
[27] leenhardt a,
lucey v,
denjoy i,
grau f,
do ngoc d,
coumel p. Catecholaminergic polymorphic ventricular tachycardia in children: a 7-year follow-up of 21 patients.
Circulation. 1995;
91:1512
-1519
[28] lupoglazoff jm,
denjoy i,
guicheney p,
casasoprana a,
coumel p. Syndrome du QT long congénital.
Arch Pédiatr. 2001;
8:525
-534
[29] lupoglazoff jm,
denjoy i,
villain e,
fressart v,
legall-petit i, et coll.. Neonatal forms of congenital long QT syndrome.
Arch Mal Coeur Vaiss. 2004;
97:479
-483
[30] maron bj,
gardin jm,
flack jm,
gidding ss,
kurosaki tt,
bild de. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults.
Circulation. 1995a;
92:785
-789
[31] maron bj,
pelliccia a,
spirito p. Cardiac disease in young trained athletes. Insights into methods for distinguishing athlete’s heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy.
Circulation. 1995b;
91:1596
-1601
[32] mckoy g,
protonotarios n,
crosby a,
tsatsopoulou a,
anastasakis a, et coll.. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease).
Lancet. 2000;
355:2119
-2124
[33] mc nair wp,
ku l,
taylor mr,
fain pr,
dao d, et coll.. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia.
Circulation. 2004;
110:2163
-2167
[34] meune c,
van berlo jh,
anselme f,
bonne g,
pinto ym,
duboc d. Primary prevention of sudden death in patients with lamin A/C gene mutations.
N Engl J Med. 2006;
354:209
-210
[35] mohler pj,
bennett v. Ankyrin-based cardiac arrhythmias: a new class of chan-nelopathies due to loss of cellular targeting.
Curr Opin Cardiol. 2005;
20:189
-193
[36] moss aj,
schwartz pj,
crampton rs,
tzivoni d,
locati eh, et coll.. The long QT syndrome: prospective longitudinal study of 328 families.
Circulation. 1991;
84:1136
-1144
[37] norgett ee,
hatsell sj,
carvajal-huerta l,
cabezas jc,
common j, et coll.. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma.
Hum Mol Genet. 2000;
9:2761
-2766
[38] postma av,
denjoy i,
hoorntje tm,
lupoglazoff jm,
da costa a, et coll.. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia.
Circ Res. 2002;
91:e21
-e26
[39] postma av,
denjoy i,
kamblock j,
alders m,
lupoglazoff jm, et coll.. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients.
J Med Genet. 2005;
42:863
-870
[40] priori sg,
napolitano c,
tiso n,
memmi m,
vignati g, et coll.. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia.
Circulation. 2000;
102:r49
-r53
[41] rampazzo a,
nava a,
malacrida s,
beffagna g,
bauce b, et coll.. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy.
Am J Hum Genet. 2002;
71:1200
-1206
[42] richard p,
charron p,
carrier l,
ledeuil c,
cheav t, et coll.. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy.
Circulation. 2003;
107:2227
-2232
[43] romano c,
gemme g,
pongiglione r. Aritmie cardiache rare dell’eta pediatria.
Clin Pediatr. 1963;
45:656
-683
[44] royer a,
van veen ta,
le bouter s,
marionneau c,
griol-charhbili v, et coll.. Mouse model of SCN5A-linked hereditary Lenegre’s disease: age-related conduction slowing and myocardial fibrosis.
Circulation. 2005;
111:1738
-1746
[45] saffitz je. Structural heart disease, SCN5A gene mutations, and Brugada syndrome: a complex menage a trois.
Circulation. 2005;
112:3672
-3674
[46] schott jj,
alshinawi c,
kyndt f,
probst v,
hoorntje tm, et coll.. Cardiac conduction defects associate with mutations in SCN5A.
Nat Genet. 1999;
23:20
-21
[47] schulze-bahr e. Short QT syndrome or Andersen syndrome: Yin and Yang of Kir2.1 channel dysfunction.
Circ Res. 2005;
96:703
-704
[48] seidman jg,
seidman c. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms.
Cell. 2001;
104:557
-567
[49] sen-chowdhry s,
syrris pw. Genetics of right ventricular cardiomyopathy.
J Cardiovasc Electrophysiol. 2005;
16:927
-935
[50] splawski i,
shen j,
timothy k,
lehmann ml,
priori s, et coll.. Spectrum of muta-tions in long QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2.
Circulation. 2000;
102:1178
-1185
[51] tiso n,
stephan da,
nava a,
bagattin a,
devaney jm, et coll.. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhyth-mogenic right ventricular cardiomyopathy type 2 (ARVD2).
Hum Mol Genet. 2001;
10:189
-194
[52] viswanathan pc,
balser jr. Inherited sodium channelopathies: a continuum of channel dysfunction.
Trends Cardiovasc Med. 2004;
14:28
-35
[53] ward oc. A New familial cardiac syndrome in children.
J Irish Med Assoc. 1964;
54:103
-106
[54] wilde aa,
antzelevitch c,
borggrefe m,
brugada j,
brugada r, et coll.. Proposed diagnostic criteria for the Brugada syndrome: consensus report.
Circulation. 2002;
106:2514
-2519
Pascale Guicheney
Institut de myologie, Inserm U 582
Groupe hospitalier Pitié-Salpêtrière, Paris
Tests présymptomatiques en neurogénétique
Un des faits nouveaux en génétique médicale est d’avoir montré que les maladies héréditaires à révélation tardive sont plus nombreuses et plus fréquentes que l’on croyait. On peut estimer qu’environ 1 %, peut-être plus, de la population d’âge adulte est concerné par ces pathologies. Différentes maladies, la plupart de nature neurodégénérative, sont concernées. De nombreux gènes sont déjà identifiés et l’hétérogénéité génétique s’est révélée très importante. Ces connaissances génétiques permettent donc un diagnostic présymptomatique par analyse moléculaire chez le sujet à risque élevé pour ces affections.
Certaines de ces maladies sont monogéniques dans la totalité ou la quasi-totalité des cas, telles la maladie de Huntington ou la dystrophie myotonique de Steinert ; d’autres au contraire sont des sous-entités mendéliennes de maladies qui, dans la grande majorité des cas seront multifactorielles, citons les démences de type Alzheimer, ou la maladie de Parkinson (tableau I
). Il est cependant troublant de noter que dans le cas de la maladie de Parkinson les formes héréditaires monogéniques ne se distinguent pas des formes dites « sporadiques ou idiopathiques » de la même maladie, autant en ce qui concerne l’âge de début de la maladie et la progression. Ces maladies se transmettent dans la majorité des cas selon un mode dominant autosomique, parfois avec une pénétrance réduite. Certaines sont très graves, aucun traitement préventif ou curatif n’existe ; d’autres peuvent être traitées, ou prévenues avec une efficacité variable selon la maladie.
De façon schématique, un test génétique peut être proposé dans trois situations différentes. La première est de confirmer le diagnostic d’une maladie génétique chez une personne ayant des symptômes ; le test peut alors être considéré comme un moyen de porter avec certitude un diagnostic évoqué cliniquement. La deuxième situation concerne les maladies neurologiques à révélation tardive ; elles peuvent susciter une demande de diagnostic présymptomatique, « faire un test génétique » avant que des signes de la maladie héréditaire soient apparus. Être porteur, développer la maladie et risquer de la transmettre, ou ne pas être porteur et ne pas transmettre la maladie sont les deux résultats possibles dans cette deuxième situation. La troisième situation est le diagnostic prénatal pour un fœtus à risque, acte controversé et difficile dans les maladies qui se manifestent à l’âge adulte. Nous centrerons cette revue sur les tests présymptomatiques et leurs conséquences sur les demandes de diagnostic prénatal.
Interrogations face au test présymptomatique
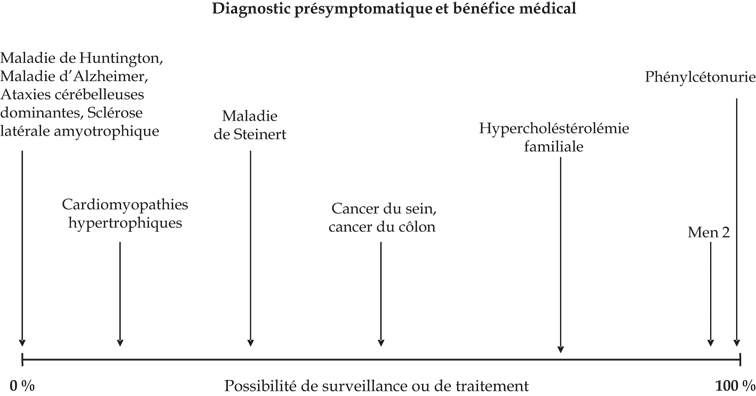
Un premier niveau de la réflexion, « faire ou ne pas faire un test prédictif », s’organise autour des possibilités de traitement ou de prévention. Il serait idéal de transformer un test présymptomatique en test de « dépistage », terme qui implique un traitement ou une prévention comme conséquence immédiate du test. La question de faire ou de ne pas faire serait obsolète. Les possibilités de prévention s’étalent entre le dépistage, c’est-à-dire la possibilité d’une prévention maximale (par exemple, chirurgie préventive dans le cas des cancers de la thyroïde dans la néoplasie endocrine multiple de type II), et le test « brut » sans possibilité de traitement et de prévention après un résultat défavorable, dans le cas de maladies neurodégénératives dont l’exemple le plus connu est celui de la maladie de Huntington, mais d’autres sont cités dans le tableau I. Entre ces deux extrêmes, les maladies s’échelonnent avec une possibilité de prévention ou de traitement plus ou moins efficaces (surveillance renforcée voire mastectomie et ovariectomie dans le cancer du sein dû à une mutation BRCA1). Selon la pathologie, le degré d’intervention possible varie et ceci est schématiquement présenté dans la figure 1
Tableau I Exemples de maladies neurodégénératives héréditaires pour lesquelles existent des formes héréditaires et pour lesquelles un test prédictif est possible dans certains sous-groupes génétiques
|
Maladie
|
Forme(s)
monogénique(s)
Prévalence en Europe
|
Principaux gènes impliqués
|
Formes non
génétiques
|
|
Maladies de Huntington
|
0,5-1/10 000
|
IT15 (90 %), HDL2 (< 1 %), PrP (< 1 %)
|
–
|
|
Maladie d’Alzheimer
|
1/10 000
|
APP, PSEN1, PSEN2
|
+++
|
|
Maladie de Creutzfeld-Jakob
|
< 1/100 000
|
PrP
|
+
|
|
Maladie de Parkinson
|
1/5 000
|
α-synuclein, Parkin, UCHL-1, Pink1, DJ1, LRRK2
|
+++
|
|
Sclérose latérale amyotrophique
|
Oui
|
SOD1
|
++
|
|
Ataxies cérébelleuses autosomiques dominantes
|
3-5/100 000
|
SCA1-27
|
–
|
|
Paraparésies spastiques autosomiques dominantes
|
2-3/100 000 ?
|
SPG3, 4, 6, 8, 9, 10, 12, 13, 17, 19
|
–
|
|
Myotonie de Steinert
|
1/25 000
|
DM1, DM2
|
–
|
–, +, ++, +++ : proportion de formes non génétiques (pourcentages exacts difficiles à estimer)
La prise de décision chez la personne à risque prendra en compte les possibilités de prévention et le vécu de la maladie chez le membre atteint de la famille. En effet, une maladie jugée relativement bénigne pour les médecins mais associée à un vécu douloureux individuel et familial peut être à l’origine de demandes de test présymptomatique et même prénatal.
Pouvoir prédictif du test présymptomatique et ses limites
Il s’agit du deuxième niveau de discussion autour de la question de faire ou de ne pas faire un test prédictif. Selon la maladie, le pouvoir prédictif du test est très différent. A priori, la valeur prédictive est claire pour les maladies monogéniques avec une pénétrance complète : si vous êtes porteur, vous allez développer la maladie. Mais quand et comment ? L’âge de début de la maladie est souvent très variable pour les maladies de transmission dominante, même à révélation tardive : en moyenne entre 30 et 50 ans mais des formes après 60 ans ne sont pas rares. La pénétrance incomplète de la mutation et l’expressivité clinique variable de la maladie sont à la base du faible pouvoir prédictif pour beaucoup de maladies dominantes, à révélation tardive. Le test génétique ne permet donc pas réellement de prédire précisément l’avenir de la personne porteuse. Quels seront l’âge de début, l’évolution et la gravité ? Le test génétique détermine un génotype mais non pas un phénotype. Ainsi, le pouvoir prédictif du test génétique est à la fois fort car il donne une quasi-certitude d’être atteint en cas de résultat défavorable, mais il est aussi faible car il est incapable de prédire le début et l’évolution.
Dans les maladies multifactorielles qui relèvent tout à la fois de facteurs génétiques et de facteurs environnementaux, le pouvoir prédictif du test génétique est très faible. En effet, les facteurs génétiques impliqués sont largement répandus dans la population générale et bon nombre de personnes porteuses d’un de ces facteurs ne développeront jamais la maladie. Dans le cas de la maladie d’Alzheimer, l’allèle E4 de l’apolipoprotéine (ApoE4) est un facteur de risque important avec un risque augmenté d’un facteur de 2 à 3 chez les hétérozygotes et de 7 à 9 chez les homozygotes. Le test génétique est peu contributif pour confirmer la maladie chez une personne malade, puisque d’authentiques patients avec une maladie d’Alzheimer ne portent pas l’allèle E4. Pour l’instant, le test génétique et la révélation de ce facteur de risque n’ont aucune utilité clinique chez une personne saine.
La prédiction est donc très relative, il s’agit plus de la révélation d’un statut génétique que de l’état clinique et les personnes à risque doivent connaître les incertitudes médicales quant à la prédiction per se.
Le diagnostic présymptomatique ne se conçoit donc actuellement que chez une personne à risque pour une affection monogénique qui souhaite réellement connaître son statut génétique après le diagnostic de l’affection chez un malade dans la famille, sans en tirer un bénéfice médical immédiat et en acceptant les incertitudes quant aux détails cliniques.
Exemple de la maladie de Huntington
La maladie de Huntington (MH) a donné lieu à la réflexion la plus approfondie et bénéficie du plus grand recul dans la pratique de tests présymptomatiques. Cette maladie est la première pour laquelle un test présymptomatique devint techniquement possible par une analyse indirecte dès la fin des années 1980 et fiable, par analyse directe, depuis 1993 avec la découverte du gène responsable de la maladie et de sa mutation causale. C’est une maladie de transmission autosomique dominante et c’est par ailleurs une histoire familiale de troubles psychiatriques, cognitifs et de comportement ou mouvements anormaux qui est très souvent retrouvée. Il s’agit d’une maladie héréditaire rare avec une prévalence estimée à 0,5 à 1/10 000 en Europe et qui touche toutes les ethnies (Bates et coll., 2002
). La base moléculaire de la maladie de Huntington est l’expansion anormale d’un trinucléotide CAG (cytosine, adénine, guanine) dans le premier exon du gène
IT15 codant la huntingtine ; ce gène est localisé sur le bras court du chromosome 4. Le nombre de répétitions est inférieur à 30 sur les chromosomes normaux et supérieur ou égal à 36 chez les patients. Dans la majorité des cas, la longueur de l’expansion se situe entre 40 et 45 CAG. Il existe une corrélation inverse entre le nombre de CAG et l’âge de début, la sévérité clinique et l’intensité des lésions neuropathologiques mais il existe d’importantes variations individuelles. Plus le nombre de répétitions est grand, plus l’âge de début est précoce. Néanmoins, le nombre de répétitions CAG ne permet pas de prédire précisément l’âge de début ou la rapidité d’aggravation de la maladie chez un patient donné. Dans la plupart des cas, l’instabilité de la répétition pendant la transmission a pour conséquence l’augmentation de quelques unités du nombre des triplets CAG chez l’enfant porteur. Dans quelques cas rares, la transmission paternelle s’accompagne d’une grande augmentation de la longueur de l’expansion, qui peut dépasser alors 60 répétitions CAG, et être à l’origine de cas juvéniles de la maladie.
L’hétérogénéité génétique est prouvée depuis la découverte d’un second gène
JPH3 (Holmes et coll., 2001
). Le phénotype de la MH due à une mutation dans
IT15 ou
JPH3 ne se distingue pas cliniquement mais il a été montré que
JPH3 est beaucoup plus fréquemment associé à une MH chez les patients d’origine africaine (30 % en Afrique du Sud parmi les formes cliniquement établies de MH ; Krause et coll., 2002
). De plus, les formes adultes de l’atrophie dentato-rubropallido-luysienne (DRPLA), beaucoup plus fréquentes au Japon qu’en Europe, peuvent mimer une maladie de Huntington.
En France, la mise en place d’une structure d’accueil pour le test présymptomatique de la maladie de Huntington dès 1992 permet aujourd’hui de connaître mieux les motivations et les souhaits des demandeurs, les conséquences de la prédiction sur leur vie et la descendance et l’intérêt d’une structure de prise en charge multidisciplinaire.
Une réflexion internationale a conduit à l’élaboration de règles qui encadrent les bonnes pratiques du diagnostic présymptomatique de la maladie de Huntington (
International Huntington Association et
World Federation of Neurology, 1994
). Cinq principes sont mis en avant : bénéfice, autonomie, choix éclairé, confidentialité, et égalité. Dans ce cadre, le bénéfice n’est pas thérapeutique et dépend de la demande individuelle de la personne à risque au test. Le principe d’autonomie requiert que le test ne soit demandé qu’à titre individuel et par une personne majeure. Le choix éclairé nécessite de délivrer une information aussi complète que possible sur la maladie et ses caractéristiques génétiques ainsi que les différentes options en matière de test. La confidentialité est capitale pour l’avenir de la personne à risque, surtout si elle reçoit une réponse défavorable. Enfin, le principe d’égalité s’applique aux possibilités d’accès de la personne à risque aux centres qui pratiquent le test présymptomatique sans discrimination de nature financière. Il est clair qu’un diagnostic présymptomatique n’est utile que s’il est à l’origine d’une prise en charge médicale et psychologique.
Afin de prendre en compte les exigences du choix informé, le déroulement du test présymptomatique dans le temps et la prise en charge de la personne à risque par une équipe pluridisciplinaire sont particulièrement importants. La composition de l’équipe reflète les problèmes soulevés par la maladie concernée : généticien et neurologue connaissant la maladie ; psychologue averti de la transmission autosomique dominante et du devenir des personnes à risque après le test ; assistante sociale au courant des enjeux sociaux et des conséquences dans le domaine des assurances ; et infirmière de génétique consciente de la nécessité du déroulement des entretiens dans le temps et de la disponibilité de l’équipe. L’existence de plusieurs interlocuteurs va permettre une réflexion variée en raison de leurs approches différentes de la maladie, afin de prendre en compte la spécificité de chaque demande. Il est intéressant de constater que le désir de savoir est le moteur le plus grand pour faire le test et que même une grossesse en cours n’est pas plus incitative à faire un test (Lesca et coll., 2002
).
Un cadre de trois entretiens après le premier contact jusqu’à la prise de décision et le prélèvement sanguin est proposé : une consultation avec le psychologue, une avec l’assistante sociale et une dernière avec le généticien. Les personnes à risque connaissent le nom des intervenants, une brochure leur est remise avec le numéro de téléphone direct qui permet une organisation adaptée des rendez-vous successifs. La possibilité de rencontrer un psychiatre ou faire un test de mémoire, pour détecter les premiers signes de la maladie, est également offerte. La consultation a été mise en place dès 1992 à l’Hôpital de la Salpêtrière. Aujourd’hui, 15 centres nationaux prennent en charge les demandes.
Depuis la mise en place de notre centre
1
, 984 personnes à risque sont venues à cette consultation pour demander un test présymptomatique, 527 (55 %) ont continué la démarche en rencontrant l’ensemble des membres de la consultation, dont 352 ont obtenu le résultat. Trois chiffres soulignent l’importance du temps dans la réflexion de la personne à risque : seules 20 % des personnes à risque formulent une demande de test ; une personne sur deux ne poursuit pas sa démarche après le premier entretien ; une personne sur dix choisit de suspendre sa démarche entre le premier entretien et la prise de sang (Goizet et coll., 2002
).
Existe-t-il une différence entre ceux qui poursuivent et ceux qui ne souhaitent pas aller jusqu’au bout de leur demande de diagnostic présymptomatique ? Une étude réalisée dans notre équipe a montré que ceux qui poursuivent ont plusieurs motivations et sont plus orientés vers un projet parental, ou ont déjà des enfants et souhaitent informer leurs enfants de leur risque. En revanche, ceux qui abandonnent ont une connaissance récente de leur risque et un nombre plus limité de motivations. C’est dans cet esprit que le décret du 23 juin 2000 limite la prescription des tests présymptomatiques à l’intervention d’équipes pluridisciplinaires, rassemblant les compétences médicales nécessaires : « Chez une personne asymptomatique, mais présentant des antécédents familiaux, la prescription d’un examen des caractéristiques génétiques ne peut avoir lieu que dans le cadre d’une consultation médicale individuelle. Cette consultation doit être effectuée par un médecin œuvrant au sein d’une équipe pluridisciplinaire rassemblant des compétences cliniques et génétiques. Cette équipe doit se doter d’un protocole type de prise en charge et être déclarée au Ministère chargé de la santé… » (Art. 145-15-5). Dans le même décret, la réalisation de ce test chez un mineur est interdite, sauf si ce dernier ou sa famille peut bénéficier personnellement de mesures préventives ou curatives immédiates.
Le test présymptomatique est loin d’être un acte médical neutre. Le résultat équivaut soit à une condamnation, soit à une libération. Quel est l’impact du résultat et les conséquences d’un résultat défavorable ?
Dans la maladie de Huntington, une étude multicentrique montre que, dans ces structures, la proportion d’événements indésirables graves en rapport avec le test présymptomatique reste limitée que le résultat soit défavorable ou non (Almqvist et coll., 1999
). Les porteurs sont plus souvent déprimés (56 %) et vont moins bien que les non-porteurs. Néanmoins, 31% des non-porteurs sont déprimés aussi et nécessitent un suivi. Ceci souligne que les entretiens avec des intervenants différents, respectueux du choix de la personne à risque, jouent un rôle crucial dans la préparation au résultat et au suivi ultérieur. Même après un résultat favorable, le temps pour guérir du fait d’être à risque est important à prendre en considération (Gargiulo, 1999
). Le temps pour « guérir » du risque est individuel et il est très clairement plus court pour le conjoint que pour la personne à risque dont l’état « d’être à risque » fait partie de son identité.
Des réactions paradoxales peuvent se produire chez les non-porteurs du gène après l’annonce du résultat. Le remaniement identitaire est important, car certaines personnes à risque avaient construit toute leur vie en fonction de l’idée d’être porteurs. D’autres développent une culpabilité du survivant, par rapport à d’autres membres de la fratrie. L’annonce d’un résultat favorable ne permet pas une récupération psychologique immédiate. Il faut du temps pour assimiler cette information, qui remet parfois en question tout un projet de vie.
Le diagnostic prénatal n’est pas une conséquence directe du test présymptomatique. Ceci a été montré par les équipes canadiennes et plus récemment par un groupe de travail européen (Evers-Kiebooms et coll., 2002a
). Le diagnostic prénatal dans les maladies récessives graves se justifie non seulement par le fait que le risque pour la descendance n’est que 25 % mais aussi parce que les parents ne seront pas atteints de la maladie. En revanche, la situation est radicalement différente dans le cas des maladies dominantes car le parent transmetteur va être malade et statistiquement un fœtus sur 2 sera porteur conduisant à une interruption de grossesse. Ceci change la vision de faire naître un enfant dans un monde avec un parent malade. Enfin, plus de femmes que d’hommes sont demandeuses du test présymptomatique et donc plus de futures mères sont concernées par le fait de savoir qu’elles vont devenir malades et de transmettre la maladie. Le nombre de demandes du test présymptomatique a eu tendance à augmenter après la découverte du gène responsable mais les demandes de diagnostic prénatal après un test défavorable sont restées faibles. En effet, parmi les porteurs européens, 85 % n’ont pas eu de grossesse suite au résultat comparé aux 72 % des non-porteurs (Evers-Kiebooms et coll., 2002b
). Parmi les porteurs qui ont eu des grossesses, 60 % ont demandé un diagnostic prénatal. Il faut savoir que l’âge moyen de demande de diagnostic présymptomatique est 30 ans ce qui signifie que la majorité des personnes qui demandent un diagnostic pour elles-mêmes ont déjà des enfants. Il a été montré également qu’une grossesse en cours ne conduit pas automatiquement à la réalisation d’un test prénatal (Lesca et coll., 2002
).
Autres maladies neurodégénératives pour lesquelles un test présymptomatique est possible
Les personnes à risque pour d’autres maladies neurodégénératives peuvent maintenant demander un test présymptomatique (tableau I
). Cependant, les gènes impliqués sont connus depuis moins longtemps et certaines de ces pathologies sont très rares d’où une expérience plus limitée. Notre expérience concerne surtout les ataxies cérébelleuses autosomiques dominantes causées par les mutations des gènes SCA (
spinocerebellar ataxia) 1, 2, 3, 6 et 7 pour lesquelles des différences avec la maladie de Huntington sont mises en évidence. En termes de motivations, les personnes à risque pour une ataxie cérébelleuse dominante mettent en avant leur crainte d’être atteintes, rare chez les personnes à risque pour la maladie de Huntington (24 % versus 7 %). En revanche, le désir de savoir est plus grand chez les personnes à risque pour la maladie de Huntington (57 % versus 35 %), de même que le souhait d’informer les enfants (25 % versus 8 %) (Goizet et coll., 2002
). Ceci souligne la difficulté supplémentaire des personnes à risque dans la maladie de Huntington, du fait qu’elles savent qu’elles ne vont pas se rendre compte d’un début de la maladie en raison de l’anosognosie inhérente à l’affection, très différente d’un déni actif. Les personnes à risque pour une ataxie cérébelleuse dominante sont au contraire les premières à s’apercevoir du début des troubles alors que dans la maladie de Huntington, c’est l’entourage. D’autres différences sont notables. Ainsi, le problème de la transmission à la descendance est plus important dans les SCA que dans la maladie de Huntington. Ce fait est à rapprocher de l’observation que les grossesses chez un couple porteur s’accompagnent beaucoup plus souvent d’une demande de diagnostic prénatal dans les SCA que dans la maladie de Huntington. Néanmoins, ces différences de motivations entre les groupes de personnes à risque n’empêchent pas la survenue d’événements indésirables (dépression ou autres) après le résultat du test pour ces deux pathologies. Ceci souligne l’intérêt de la prise en charge pluridisciplinaire au long cours pour toutes ces maladies bien qu’elles présentent des particularités dans leurs manifestations (présence de troubles du comportement ou non) et de leur progression (perte d’autonomie ou décès rapide versus maladie à progression lente sans retentissement sur l’espérance de vie).
D’autres questions seront abordées avec de nouvelles pathologies. Par exemple dans le cas de la maladie de Huntington ou des SCA, la pénétrance est complète impliquant qu’être porteur signifie obligatoirement développer la maladie. Dans des paraplégies spastiques autosomiques dominantes (SPG4, par exemple) ou avec certaines mutations de la maladie de Creutzfeld-Jakob (E200K, par exemple), la pénétrance est incomplète. Le fait de pouvoir encore échapper à la maladie après un résultat défavorable a forcément une influence sur la décision de faire ou non le test présymptomatique.
En conclusion, la consultation de diagnostic présymptomatique doit être réalisée par une équipe pluridisciplinaire accueillant le demandeur dans le respect de la temporalité de la prise de décision et d’une annonce difficile. L’objectif d’éclairer une personne sur son statut génétique sans pouvoir lui proposer une prévention ou de traitement doit être mené sans précipitation.
Bibliographie
[1] almqvist ew,
bloch m,
brinkman r,
craufurd d,
hayden mr, et coll.. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease.
Am J Hum Genet. 1999;
64:1293
-1304
[2] bates g,
harper ps,
jones ps. Huntington’s disease.
3
rd ed. Oxford University Press,;
Oxford: UK.
2002;
2861
[3] evers-kiebooms g,
zoeteweij mw,
harper ps. Prenatal testing for late onset neu-rogenetic diseases.
In: evers-kiebooms g, zoeteweij mw, harper ps (eds), editors.
Bios Scientific Publishers Limited;
Royame-Uni.
2002a;
219 pp.
[4] evers-kiebooms g,
nys k,
harper ps,
zoeteweij m,
durr a, et coll.. Predictive DNA-testing for Huntington’s disease and reproductive decision making: a European collaborative study.
Eur J Hum Genet. 2002b;
10:167
-176
[5] gargiulo m. Guérir du risque ? Numéro spécial médecine prédictive. Quelle place pour l’homme ?.
Revue Laenec Médecine Santé Ethique. 1999;
3-4:16
-19
[6] goizet c,
lesca g,
durr a,
the french group for presymptomatic testing in neurogenetic disorders . Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias.
Neurology. 2002;
59:1330
-1336
[7] holmes se,
o’hearn e,
rosenblatt a,
callahan c,
hwang hs, et coll.. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2.
Nat Genetics. 2001;
29:377
-378
[8]international huntington association, world federation of neurology. Guidelines for the molecular genetics predictive tests in Huntington disease.
Neurology. 1994;
44:1533
-1536
[9] krause a,
temlett j,
van der meyden k,
ross ca,
callahan c,
margolis rl. CAG/CTG repeat expansions at the HDL2 locus are a common cause of Huntington disease in Black South Africans.
Am J Hum Genet. 2002;
71:528
[10] lesca g,
goizet c,
durr a,
the french group for presymptomatic testing in neurogenetic disorders . Predictive testing in the context of pregnancy: expe-rience in Huntington’s disease and autosomal dominant cerebellar ataxia.
J Med Genet. 2002;
39:522
-525
Alexandra Dürr
Département de génétique cytogénétique et embryologie
Groupe hospitalier Pitié-Salpêtrière, Paris
Pratique des tests génétiques en médecine
L’irruption de la biologie moléculaire en médecine au début des années 1980 a inauguré l’ère de la médecine moléculaire. En ce qui concerne les maladies génétiques, une pratique biologique entièrement nouvelle est née, la génétique moléculaire médicale. Celle-ci est fondée sur la connaissance des gènes de maladie, et plus généralement du répertoire de l’ensemble des gènes humains et du génome, dont la séquence complète est maintenant connue. Elle a pour objet d’assurer des diagnostics génotypiques fondés sur la caractérisation des anomalies du génome responsables de la pathologie (essentiellement analyse des gènes, et par extension de leurs produits d’expression : ARN messager et in fine protéine).
Elle est caractérisée par les éléments suivants :
• elle réclame des compétences théoriques et un savoir-faire très spécialisés, ce qui a conduit en particulier à créer en 2003 une spécialisation génétique au sein du DES de biologie médicale (décret n° 2003-76 du 23 janvier 2003 et arrêtés du 4 juillet 2003 et du 13 avril 2006) ;
• elle repose sur des concepts spécifiques (distincts de la biologie classique) et des techniques nouvelles et évolutives ;
• elle réclame une veille permanente en raison des progrès très rapides dans la connaissance du génome humain et de sa pathologie ;
• elle implique une participation active des praticiens aux recherches dans ce domaine, car elle se situe à la charnière entre les découvertes de la recherche fondamentale et leur application au diagnostic (activité de transfert) ;
• elle intègre la participation du médecin de famille, du spécialiste, du généticien clinique ;
• elle confère à ses acteurs une responsabilité sans précédent en biologie clinique, puisqu’elle débouche sur un diagnostic définitif
1
d’une maladie déclarée (diagnostic positif) ou à venir (diagnostic prédictif). Elle éclaire le clinicien sur la cause de la maladie, et parfois sur son pronostic. Cette activité est donc bien différente de l’activité semi-industrielle du plateau technique traditionnel ;
• elle est assortie d’implications éthiques majeures.
L’impact médical des progrès accomplis au cours des 30 dernières années dans la compréhension du déterminisme génétique des maladies est considérable et déborde le strict cadre des maladies déjà réputées génétiques en raison de leur caractère héréditaire évident. En plus du cancer qui est une pathologie à la fois héréditaire et somatique de l’ADN, un grand nombre de syndromes, non considérés jusqu’ici comme du ressort de la pathologie génétique en raison de leur caractère sporadique, apparaissent à présent comme dus à une pathologie monogénique (nombreuses formes de cécités, de surdités, de maladies neuro-dégénératives, de cardiomyopathies « idiopathiques », de cancers...).
Sur un plan cognitif, l’accès aux gènes aura aussi ouvert la voie de la compréhension des grandes énigmes biologiques (origine de la vie, évolution, spéciation, embryogenèse, morphogenèse, différenciation, organisation du système nerveux...), avec une application immédiate à la médecine (par exemple la découverte des nombreux gènes intervenant dans le développement de l’embryon des mammifères et de leurs équivalents chez l’homme va rendre intelligible la pathologie embryo-foetale).
Trois grandes leçons se dégagent en matière de pathologie moléculaire humaine :
• l’hétérogénéité génétique : une même maladie, qui jusqu’alors apparaissait comme univoque aux yeux des cliniciens, peut être en fait due à l’atteinte alternative de plusieurs gènes (exemples : les rétinites pigmentaires, les myocardiopathies hypertrophiques, les myopathies des ceintures). Autrement dit « une maladie-plusieurs gènes » ;
• l’hétérogénéité allélique : des mutations différentes dans un même gène peuvent induire des maladies différentes (exemples : le gène du récepteur aux androgènes dont certaines lésions donnent une insensibilité aux androgènes, et d’autres lésions une maladie neurologique ; le gène CFTR dont la plupart des lésions entraînent une mucoviscidose classique et certaines une stérilité masculine isolée). Autrement dit « un gène-plusieurs maladies » ;
• la spécificité de la pathologie de chaque gène : certains gènes sont le siège de mutations de type délétion, d’autres sont plutôt le siège de mutations ponctuelles ; certains présentent des anomalies peu ou pas diversifiées (exemple : la drépanocytose), d’autres au contraire sont remarquables par la variété et la dispersion des mutations tout au long du gène (exemple : le gène BRCA1 impliqué dans la prédisposition génétique au cancer du sein et/ou de l’ovaire).
Ces particularités ont deux conséquences :
• elles bouleversent la nosologie anatomo-clinique traditionnelle ;
• elles compliquent singulièrement le diagnostic moléculaire, puisqu’elles réclament une stratégie d’analyse génotypique propre à chaque gène et à chaque maladie.
Les problèmes posés en France par la génétique moléculaire médicale, avec un recul d’environ 20 ans, concernent son organisation au plan national, (meilleure visibilité des réseaux existants, articulation avec les différentes activités médicales, place dans le cadre de la génétique médicale, organisation des contrôles de qualité, reconnaissance de centres de référence) ; son développement rapide compte tenu des progrès spectaculaires de la génomique, tant sur le plan cognitif que méthodologique ; son financement (NABM : nomenclature des actes de biologie moléculaire, totalement inadaptée, problèmes de prise en charge, inadéquation au mode de financement de la santé, facturation des actes). C’est une « affaire de santé publique ».
Organisation de la génétique moléculaire en France
La génétique moléculaire s’est mise en place en France très progressivement à partir de 1985 pour satisfaire des besoins nouveaux, nés des premiers résultats obtenus dans la caractérisation des gènes de maladies et de leur pathologie. Cette mise en place initiale s’est exclusivement effectuée grâce à des initiatives individuelles de certains biologistes impliqués dans ces progrès, souvent soutenus par des associations de malades. Devant l’indifférence et l’absence totale d’investissement des pouvoirs publics, malgré des enjeux majeurs, les praticiens de génétique moléculaire se sont regroupés et ont créé en 1996 une association : l’ANPGM (Association nationale des praticiens de génétique moléculaire) considérant à cette époque que le moment était venu de dresser un bilan, d’exposer les difficultés rencontrées et prévisibles, et de proposer des remèdes. L’ANPGM a ainsi rédigé, en 1998, un livre blanc qui s’adressait essentiellement aux pouvoirs publics, aux acteurs de la santé publique, et, par delà, aux malades concernés. Il est resté sans écho jusqu’en 2001 où, sous la pression des associations de malades, les pouvoirs publics se sont enfin intéressés au problème et ont permis la concrétisation des grandes lignes du livre blanc pour conduire à une organisation en réseaux qui sera décrite plus loin. Ces réseaux ont obtenu des financements pérennes à la suite d’appels d’offres successifs de la Direction de l’hospitalisation et de l’organisation des soins (Dhos). La conséquence est que seuls les laboratoires des hôpitaux publics ont reçu une aide pour réaliser l’activité de génétique moléculaire. Dans la pratique, les laboratoires privés sont complètement exclus, puisqu’ils n’ont pas reçu de financements spécifiques et que les actes correspondants ne sont pas inscrits à la NABM.
À l’heure actuelle, la pratique de la génétique médicale est encadrée par des textes officiels : Loi de Bioéthique (juillet 1994). Cette loi a été révisée en août 2004, certains décrets d’application sont encore attendus, notamment celui fixant les conditions de prescription et de réalisation de l’examen des caractéristiques génétiques d’une personne ou son identification par empreintes génétiques à des fins médicales.
Il n’y a pas de définition unique des tests génétiques. Nous nous limiterons ici à celle qui devrait être retenue pour le décret évoqué dans le paragraphe précédent. À savoir : « L’examen des caractéristiques génétiques d’une personne à des fins médicales s’entend de celui de ses caractéristiques génétiques héritées ou acquises à un stade précoce du développement prénatal. Cet examen a pour objet :
• soit de poser, de confirmer ou d’infirmer le diagnostic d’une maladie à caractère génétique chez une personne ;
• soit de rechercher les caractéristiques d’un ou plusieurs gènes susceptibles d’entraîner le développement d’une maladie chez une personne ou les membres de sa famille potentiellement concernés ;
• soit de contribuer à la prise en charge médicale d’une personne.
L’examen des caractéristiques génétiques d’une personne à des fins médicales comprend des analyses de biologie médicale qui ne peuvent être réalisées que par des laboratoires autorisés et par des praticiens ayant la responsabilité de ces analyses en application des articles L. 2131-1 à L. 2131-5 et R. 2131-1 à R. 2131-9 du Code de la santé publique.
On distingue :
• les analyses de cytogénétique, y compris les analyses de cytogénétique moléculaire ;
• les analyses de génétique moléculaire ;
• les autres analyses de biologie médicale qui sont prescrites dans l’intention d’obtenir des informations équivalentes dans la détermination des caractéristiques génétiques d’une personne à celles obtenues par les analyses mentionnées ci-dessus.
Il existe plus de 5 000 maladies génétiques, dont la majorité ne touche que quelques individus. L’organisation de la prise en charge de telles pathologies, compte tenu de leur faible fréquence, ne peut être pensée qu’à l’échelon de la nation, voire même de l’Europe, suivant le principe de subsidiarité, et non à l’échelon régional qui est celui sur lequel est fondée en France l’organisation du système sanitaire. Néanmoins, il importe d’être vigilant quant à l’évolution des systèmes de santé à l’échelle de l’Union européenne (cf. rapport d’Information du Sénat fait au nom de la délégation pour l’Union européenne sur « l’Union européenne et les services de santé – séance du 30 janvier 2007 »).
Compte tenu de l’hétérogénéité des actes réalisés par les laboratoires de génétique moléculaire médicale, un cadre unique ne peut être adopté. Deux grands types d’activités, correspondant à des schémas organisationnels différents, ont été définis.
Les actes techniquement simples et non susceptibles d’évoluer significativement à court ou moyen terme et pour lesquels il existe des trousses qui satisfont à la Directive Européenne 98/79/CE. Il s’agit par exemple de la recherche de la mutation p.C282Y du gène HFE responsable de l’hémochromatose génétique ou de la recherche des mutations fréquentes du gène CFTR responsable de la mucoviscidose. Ces actes simples devraient pouvoir entrer dans le cadre de l’activité classique des laboratoires et être inscrits à la NABM.
Les actes techniquement complexes et dont les stratégies sont évolutives. Le développement de ce type d’activité a résulté d’initiatives purement individuelles, les motivations étant en général historiques ou politiques (stratégie de développement et de rayonnement du laboratoire). Les moyens nécessaires ont été obtenus de manières très diverses : sollicitation d’associations caritatives, dérivation de moyens propres du laboratoire (moyens hospitaliers, universitaires, Inserm, CNRS...), contrats de toutes sortes... Dans un premier temps, on a pu constater une inadaptation totale de l’offre aux besoins, et s’est mis en place un système particulièrement hétéroclite et fragile car dépendant de sources de moyens aléatoires.
En 1998, l’ANPGM a proposé au ministère de la Santé une organisation fondée sur une structure en réseaux par pathologie. Chaque réseau est dévolu soit à une pathologie, si le nombre des sujets atteints ou des arguments médicaux le justifient, soit à un groupe de pathologies posant des problèmes médicaux et techniques proches. Chacun de ces réseaux est animé par un ou des laboratoires de référence.
Les missions de ces laboratoires de référence sont :
• d’animer un réseau dévolu à une pathologie ou un groupe de maladies ;
• de rédiger, en concertation avec tous les laboratoires appartenant au réseau, un document définissant les besoins à l’échelon national (ou européen) en termes qualitatifs (choix des stratégies d’analyses) et quantitatifs ; de proposer une répartition de l’activité entre les différents laboratoires en justifiant les choix ; la définition et la justification des moyens demandés pour l’année à venir ; la justification de l’utilisation des moyens obtenus l’année précédente. Ce document, régulièrement remis à jour, sera la base contractuelle des relations avec la tutelle ;
• d’assurer une veille technologique constante en transmettant l’information à l’ensemble des laboratoires du réseau ;
• d’assurer la réalisation des analyses les plus complexes non effectuées par les autres laboratoires du réseau ;
• d’assurer la fourniture d’éléments utiles aux autres laboratoires du réseau comme les sondes, les protocoles, les ADN contrôles...
• d’organiser au moins une fois par an une réunion avec tous les laboratoires membres du réseau destiné à faire le point à la fois au plan scientifique et logistique ;
• d’être l’interlocuteur de la tutelle pour tout ce qui concerne la ou les pathologies dont le réseau a la charge ;
• d’assurer un rôle de formation.
La désignation des centres de référence est effectuée par la tutelle sur avis d’une commission indépendante où siègent des experts étrangers.
Les principaux critères de sélection des centres de référence par la tutelle sont :
• posséder une compétence reconnue dans le domaine médical considéré (notoriété scientifique attestée par satisfaction aux critères classiques en ce domaine : publications, désignation comme expert dans les instances internationales...) ;
• avoir une activité importante dans le domaine considéré, avec un recrutement national, voire international ;
• être adossé à une structure de recherche labélisée (Inserm, CNRS, Université) ;
• s’engager à assurer la totalité des missions d’un centre de référence.
Financement
L’inscription à la nomenclature des actes de biologie médicale ne doit concerner que les actes techniquement simples, non susceptibles d’évoluer significativement à court ou moyen terme et pour lesquels il existe des trousses qui répondent de manière satisfaisante à la Directive Européenne 98/79/CE relative aux dispositifs médicaux de diagnostic in vitro. Les actes techniquement complexes et dont les stratégies sont évolutives sont réalisés au sein de laboratoires publics organisés en réseau à l’échelle du territoire et soutenus par la Dhos.
Actes techniquement simples et non susceptibles d’évoluer significativement à court ou moyen terme
Seuls les actes de génétique moléculaire constitutionnelle réalisés en vue d’un diagnostic prénatal sont actuellement inscrits à la nomenclature des actes de biologie médicale (chapitre 17, sous-chapitre 17-02 – actes de biologie moléculaire en vue du diagnostic des maladies génétiques – arrêté du 11 mars 1996). Ce sous-chapitre a plus de dix ans et mériterait d’être réactualisé.
Des propositions de modification avaient été présentées dans le cadre des travaux de la Commission consultative nationale en matière d’examens des caractéristiques génétiques à des fins médicales (cf. rapport d’activité 2000-2003, page 52). Il s’agissait en particulier de supprimer ce sous-chapitre et de le remplacer par un chapitre 18 intitulé : « Diagnostic biologique des maladies héréditaires ».
Par ailleurs, deux rapports présentés en 2003 par le Pr. Marc Delpech ont été votés par la Commission de nomenclature des actes de biologie médicale (Présidente Pr. Christiane Bébéar) :
• rapport sur l’inscription à la nomenclature des actes de biologie médicale du diagnostic génétique de la mucoviscidose par analyse du gène CFTR (Experts Dr. Emmanuelle Girodon-Boulandet et Pr. Michel Vidaud) ;
• rapport sur l’inscription à la nomenclature des actes de biologie médicale du diagnostic génétique de l’hémochromatose génétique utilisant les techniques de génétique moléculaire (Experts Pr. Véronique David et Pr. Michel Vidaud).
Ces propositions de modification de la NABM n’ont pas été suivies.
Récemment, le directeur de l’Union nationale des caisses d’Assurance Maladie (UNCAM) a décidé d’inscrire à la nomenclature des actes de biologie médicale, la recherche de la mutation p.C282Y du gène HFE1 de l’hémochromatose, suite à l’avis favorable donné pour l’inscription de cet acte par la Haute autorité de santé, en avril 2006.
La Commission de hiérarchisation des actes de biologie médicale (Présidente Pr. Christiane Bébéar) a été saisie et a donné un avis favorable à cette inscription le 11 janvier 2007. Cet acte est maintenant inscrit dans un nouveau chapitre 18 créé : « Diagnostic biologique des maladies héréditaires ». En effet, par la décision du 24 janvier 2007 relative à la liste des actes et prestations prise en charge par l’Assurance maladie, le chapitre 18 « Diagnostic biologique des maladies héréditaires » fixe les indications, dans le cadre individuel et familial, de la recherche de la mutation C282Y du gène HFE1 de l’hémochromatose.
Il est à noter que les actes de cytogénétique constitutionnelle (prénatal et post-natal) et les actes de cytogénétique moléculaire constitutionnelle sont inscrits à la nomenclature des actes de biologie médicale (chapitre 2, actes de cytogénétique, arrêté du 25 novembre 2004).
Actes techniquement complexes et dont les stratégies sont évolutives
Il est impossible pour de tels actes de déterminer une évaluation par cotation en lettre clé. En effet, le coût est variable selon les stratégies d’exploration nécessaires à développer dans les familles sans que cela puisse être prévu a priori et le nombre des examens est tellement faible qu’il est impossible de déterminer un coût forfaitaire (qui est le principe de la NABM). De plus, les techniques sont constamment évolutives. Cette impossibilité d’évaluer a priori le coût des examens et le caractère national et non régional de l’activité a conduit la Dhos à mettre en place un financement sous forme d’appels d’offres.
Des réseaux se sont progressivement mis en place à la suite des cinq appels d’offres successifs de la Dhos. Chaque année le réseau réunit tous ses membres pour faire le point, évoquer les problèmes techniques rencontrés pendant l’année et discuter les évolutions technologiques... La deuxième partie de la réunion est consacrée à la finalisation du rapport annuel du réseau qui fait un état des lieux des activités et des problèmes rencontrés. Ce rapport, adressé au Ministère, doit apporter la justification des financements attribués. Des éléments du rapport d’activité du réseau » mucoviscidose » sont présentés en annexe 2.
Tous les CHU ont obtenu des financements. Les sommes attribuées sont pérennes et ajoutées à la base budgétaire. Sans ces financements, la prise en charge des maladies rares serait restée symbolique et particulièrement inégalitaire. Les financements ont donc eu un effet bénéfique majeur. Mais de nombreux problèmes subsistent quant à l’utilisation de ces financements : le plus souvent la première année n’a jamais été créditée au budget des laboratoires ; certains CHU se sont vus refuser des recrutements, d’autres ne pouvaient utiliser une partie des crédits pour abonder des amortissements... Les mesures incitatives du ministère de la Santé n’ont pratiquement jamais fait l’objet d’un accompagnement de la part des hôpitaux.
Induits par les textes réglementaires eux-mêmes, des problèmes importants restent posés. Par exemple, dans un domaine où la technologie est en évolution continuelle, la qualité des équipements doit être en permanence adaptée. L’activité entre complètement dans le cadre de l’enveloppe Migac (Mission d’intérêt général et d’aide à la contractualisation). Comme tout examen biologique, il doit être facturé aux hôpitaux demandeurs. Nombre d’entre eux soit interdisent aux médecins l’envoi des examens au prétexte de leur coût trop élevé, soit refusent de les régler au prétexte qu’ils considèrent qu’ils ont déjà été pris en charge par le financement des réseaux attribués par la Dhos. La situation la plus catastrophique est celle où le patient a fait appel à un laboratoire privé pour se faire prélever et acheminer le prélèvement. Dans un tel cas, une facture d’un montant élevé est adressée par l’hôpital au laboratoire qui a effectué le prélèvement et celui-ci transmet la facture au patient. Il en résulte parfois, à tort, des situations de détresse inacceptables.
Pr. Michel Goossens
Président ANPGM
Pr. Marc Delpech
Vice-Président ANPGM
Pr. Michel Vidaud
Trésorier ANPGM
ANPGM
Association nationale des praticiens de génétique moléculaire
Contrôle de qualité des tests et coopération internationale
Des tests génétiques sont maintenant disponibles pour le diagnostic de plus de 1 200 maladies, la plupart rares
1
. C’est un énorme service rendu aux personnes malades et à leur famille, leur permettant de connaître avec certitude l’origine de leur maladie, son mode de transmission et les risques pour les apparentés. Cette avancée décisive pose cependant des questions dans deux domaines : celui du contrôle de qualité et celui de la coopération internationale.
En effet, le transfert maintenant très rapide de la recherche à l’utilisation clinique, est antinomique avec une stabilisation des techniques et la mise en place de standards de qualité et de mécanisme de contrôle de qualité externe. Une étude réalisée par l’OCDE publiée en 2004
2
, montrait que la participation des laboratoires à des contrôles de qualité externe était fragmentaire dans tous les pays et que peu de laboratoires de génétique moléculaire, de biochimie génétique ou de cytogénétique étaient accrédités. Des publications antérieures avaient documenté des taux d’erreurs inacceptables dans le rendu des résultats et une insuffisance d’interprétation de ceux-ci pour qu’ils soient correctement compris par les professionnels de santé et les patients. La mise en place de contrôles de qualité nécessite des moyens qui ne sont pas facilement mobilisables et une expertise qui n’est pas disponible dans tous les pays. C’est pourquoi une approche au moins européenne du sujet est indispensable. Depuis 2 ans, un projet européen, EuroGenTest, s’attache à documenter l’état d’organisation des laboratoires dans le domaine de la qualité, à organiser des réseaux de contrôles de qualité et à former les personnels. Orphanet qui liste déjà les offres de diagnostic par maladie, devrait prochainement donner accès à l’information sur l’organisation du contrôle de qualité dans chaque laboratoire avec l’espoir de promouvoir ainsi les laboratoires qui font des efforts dans ce sens. Ceci va dans le sens d’une protection des utilisateurs, cruciale dans ce secteur où beaucoup de tests sont réalisés par des laboratoires de qualité très différente et n’utilisant pas les mêmes techniques, ce qu’un professionnel non spécialiste ne peut aisément apprécier.
Le deuxième problème est celui de la coopération internationale indispensable pour le diagnostic de beaucoup de maladies rares. Un pays, même aussi grand que la France, ne peut prétendre à offrir tous les tests possibles. À l’heure actuelle, sur les 1 320 maladies génétiques pouvant être testées en Europe, seulement 917 peuvent l’être en France. Les prélèvements doivent donc voyager. Cela pose de multiples questions non résolues, en particulier les modalités de prise en charge financières de ces tests, la protection des données nominales, le contrôle de qualité de laboratoires dans des pays ayant des pratiques très différentes…
Pour les tests génétiques qui ne sont pas à visée diagnostique mais sont des outils d’évaluation du risque de survenue de maladies multifactorielles, le principal problème est l’évaluation de l’utilité clinique de ces tests. En effet, ceux-ci se diffusent actuellement sans que leur utilité n’ait été établie et commencent à peser significativement sur les budgets de santé, sans bénéfice réel aux patients. Il est intéressant de rappeler que l’enquête de l’OCDE montrait que les tests les plus pratiqués quantitativement étaient l’identification de la mutation Facteur V de Leiden et de l’APO E4 alors même qu’il n’y a pas de recommandations consensuelles sur la conduite à tenir en cas de mutation et sur le bénéfice de ce dépistage pour les personnes concernées.
Ségolène Aymé
Directrice du service information sur les maladies rares,
Inserm, Paris
Encadrement législatif et réglementaire des tests génétiques à visée médicale
La loi de bioéthique de 1994, révisée en 2004, assure un encadrement très strict de l’emploi des tests génétiques dans le champ médical (Article L. 1131-1 à -6 du CSP
1
). Sur le plan réglementaire, ces tests sont définis comme les moyens permettant « l’examen des caractéristiques génétiques d’une personne ou son identification par empreintes génétiques ». La loi prévoit que ce dispositif soit complété par des décrets d’application permettant la mise en œuvre pratique de cet encadrement (Article R. 1131-1 à -20 du CSP). Le premier décret relatif à la loi de 1994 date de 2000 et est encore en vigueur actuellement puisque le principal décret d’application de la loi de 2004 n’est pas paru. Ce dernier est en cours d’élaboration.
Globalement, le dispositif d’encadrement repose sur deux mesures principales :
• l’agrément de praticiens et l’autorisation des laboratoires qui assurent ces tests ;
• une prescription très rigoureuse avec notamment un consentement écrit des patients suivant une attestation d’information délivrée par le médecin prescrivant le test.
Agrément et autorisation
Dans le dispositif mis en place en 2000, l’agrément de praticiens et l’autorisation des laboratoires sont liés. Ils sont délivrés pour une durée de cinq ans par le préfet de région, après avis systématique de la « commission consultative nationale en matière d’examens des caractéristiques génétiques à des fins médicales » sise au ministère de la Santé (Direction générale de la santé). Ces agréments et autorisations peuvent être généraux (valables pour tous les tests génétiques) ou limités (par exemple aux facteurs de la coagulation). Il est à noter que l’agrément du praticien tombe lorsqu’il quitte la structure autorisée dans laquelle il exerce.
La loi de 2004 apporte deux changements :
• elle permet de dissocier agrément et autorisation. L’agrément est attribué pour une durée de cinq ans par le Directeur général de l’Agence de la biomédecine, sur des critères définis par le conseil d’orientation de l’établissement. L’autorisation est accordée par l’Agence régionale d’hospitalisation (ARH) à l’établissement dont dépend le laboratoire de génétique, après avis de l’Agence de la biomédecine qui lui apporte son expertise technique en la matière ;
• elle rend obligatoire la réalisation d’un bilan annuel par chaque laboratoire. L’Agence de la biomédecine est chargée de collecter ces bilans, d’en faire la synthèse et de réaliser notamment un état des lieux national.
Encadrement de la prescription des tests génétiques
Les conditions de prescription font l’objet de plusieurs mesures ayant pour finalité la meilleure information possible, d’une part des patients et d’autre part des familles concernées.
Dans le cas d’examen des caractéristiques génétiques d’une personne asymptomatique, le dispositif repose, préalablement à la signature d’un consentement éclairé par le patient, sur une attestation d’information délivrée lors d’une consultation médicale au cours de laquelle sont notamment envisagées la portée des analyses et les conséquences, tant individuelles que familiales, des résultats.
Cette consultation médicale est assurée par un praticien qui s’est déclaré auprès de la Direction générale de la santé comme appartenant à une équipe multidisciplinaire organisée pour prendre en charge ce type de pathologie.
Seul le médecin prescripteur est autorisé à rendre le résultat au patient.
La loi de 2004 propose de compléter ce dispositif par une information familiale réalisée avec l’aide de l’Agence de la biomédecine, au cas où le patient ne souhaite pas informer directement ses proches de la maladie.
En conclusion, il apparaît que ce dispositif très contraignant en matière de génétique témoigne de l’inquiétude non seulement des instances politiques mais aussi de la société en général, vis-à-vis d’éventuelles dérives en la matière.
La nécessité de transparence et d’un accord explicite laissant le choix de recourir ou non à ces tests, sont des acquis positifs pour permettre un emploi plus serein de ces techniques.
De plus, le dispositif en matière de génétique est adapté avec ce qui se fait dans le domaine prénatal, assurant ainsi une prise en charge cohérente des familles.
À terme, se posera sûrement la question de la plus-value apportée par un tel dispositif qui ne s’intéresse qu’aux laboratoires, lorsque des filières de soins plus structurées feront que la pathologie en général et celle en génétique en particulier seront prises en charge de manière intégrée et complète sous tous leurs aspects.
François Thépot
Adjoint du directeur médical et scientifique
Agence de la biomédecine