5-
Aspects génétiques de la puberté
La puberté est un indicateur de la maturation normale de l'axe gonadotrope et du système de reproduction. Ce processus physiologique survient après une période de latence dont la durée est variable en fonction du sexe puisque l'initiation pubertaire survient entre 8 et 13 ans chez la fille et entre 9 et 14 ans chez le garçon. Depuis le début du XX
e siècle, plusieurs études rétrospectives dans un premier temps et récemment prospectives, ont rapporté que l'âge de la puberté était génétiquement déterminé (Parent et coll., 2003

). Plusieurs autres facteurs ont également été impliqués tels que l'état nutritionnel, le stress, l'exercice physique (Graber et coll., 1995
). Récemment, l'évolution séculaire tendant vers une baisse continue de l'âge de la puberté, évoquée en premier par Tanner en 1973, a été expliquée par une amélioration des conditions de vie (Wyshak et Frisch, 1982
) alors que le rôle des perturbateurs endocriniens présents dans de nombreux polluants reste une question non résolue (Rogan et Ragan, 2003
).
Les travaux récents sur la génétique de la puberté poursuivent deux buts : modéliser la part des facteurs génétiques et des facteurs de l'environnement dans la puberté normale et comprendre les mécanismes des maladies de la chronologie de l'initiation de la puberté. La première approche est une approche de génétique complexe d'un trait physiologique mal défini dans le temps et souvent de façon rétrospective, ce qui pose de nombreux problèmes méthodologiques. La deuxième approche est centrée sur l'étude de maladies dites rares avec une méthodologie bien maîtrisée, concernant peu d'individus mais dont les résultats permettent des avancées majeures en physiologie et physiopathologie.
Il devient de plus en plus évident que ces deux approches sont complémentaires. Une meilleure évaluation de la part des facteurs environnementaux par rapport aux facteurs génétiques dans le déterminisme de l'âge pubertaire devenant indispensable pour mieux comprendre et évaluer la répercussion de l'environnement sur la puberté et le système de reproduction. Les nouveaux gènes décrits dans les approches de génétique monogénique mendélienne devenant des gènes candidats au déterminisme génétique de l'âge de l'initiation de la puberté normale. Ces travaux permettent de mieux comprendre les liens entre âge de l'initiation de la puberté et les pathologies dépendants de l'activation de l'axe gonadotrope telles que la baisse de la fertilité, les cancers hormono-dépendants et les maladies cardiovasculaires.
Génétique de l'âge de la puberté normale
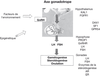
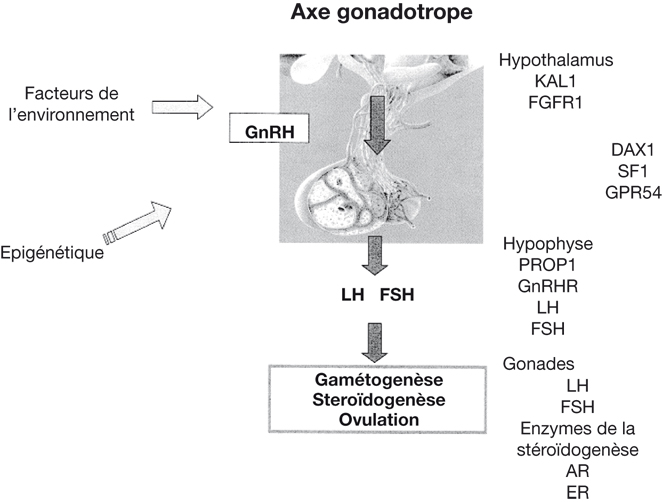
L'initiation de la puberté résulte de la maturation neuroendocrinienne de l'axe gonadotrope dont la fonction ultime est la production des hormones sexuelles et la maturation des gamètes par les gonades (Ebling, 2005
). Il est courant de diviser cet axe endocrinien en deux : la commande centrale neuroendocrinienne comprenant l'hypothalamus et l'hypophyse, et les gonades qui sont les effecteurs des hormones hypophysaires (figure 5.1
). Ces trois glandes endocrines forment un système de régulation dynamique très complexe mélangeant stimulation et rétrocontrôle négatif mais également rétrocontrôle positif. L'apparition des caractères sexuels secondaires témoigne de l'augmentation de la production des hormones sexuelles par les gonades et, par conséquent, de la réactivation de l'axe gonadotrope. L'initiation neuroendocrinienne de la puberté est antérieure à ces modifications somatiques (Manasco et coll., 1995
et 1997
). Elle est donc cliniquement muette.
Études épidémiologiques sur le déterminisme génétique de l'âge de la puberté
Pour des raisons évidentes, les études épidémiologiques sur l'âge de la puberté sont souvent réalisées chez les filles. Un interrogatoire et un examen clinique rapide permettent de dater les premières règles et de définir un stade pubertaire plus simplement que chez les garçons. Les signes les plus évidents sont le développement des seins et la ménarche. Plusieurs études, dont les premières datent de 1928, ont démontré une corrélation de l'âge de la ménarche entre sœurs, et entre mères et filles (tableau 5.I
). Cette corrélation est plus importante entre jumelles monozygotes par rapport aux dizygotes (Gedda et Brenci, 1975
; Fischbein, 1977
; Golden, 1981
). La corrélation plus importante entre jumelles dizygotes (autour de 0,6) par rapport à celle observée pour des sœurs non jumelles (autour de 0,3) est peut être le témoin d'événements partagés durant la vie fœtale (tableaux 5.I
et 5.II
). Elle pourrait également être due à un environnement postnatal similaire entre frères et sœurs du même âge par rapport à une fratrie étalée sur plusieurs années.
Tableau 5.I Héritabilité de l'âge de la ménarche : corrélations selon des études de sœurs ou de mères-filles (d'après Towne et coll., 2005)
|
Type d'études
|
Effectifs
|
Coefficient de corrélation
|
|
Références
| |
r
|
Écart-type
|
|
Études de sœurs
|
Nbre de paires de sœurs
| | |
|
Zacharias et coll., 1976
|
271
|
0,37
|
Nd
|
|
Chern et coll., 1980
| | | | |
|
Données prospectives
|
218
|
0,16
|
0,09
|
|
Données rétrospectives
|
403
|
0,25
|
0,06
|
|
Malina et coll., 1994
|
83
|
0,44
|
Nd
|
|
Études de mères et filles
|
Nbre de mères
|
Nbre de filles
| | |
|
Israel, 1959
|
≈1000
|
≈1000
|
0,28
|
Nd
|
|
Damon et coll., 1969 (d'après Bolk, 1926)
|
45
|
71
|
0,54
|
0,08
|
|
Damon et coll., 1969
|
66
|
78
|
0,24
|
0,11
|
|
Kantero et Widholm, 1971
|
nd
|
1 946
|
0,28
|
Nd
|
|
Zacharias et coll., 1976
|
577
|
577
|
0,26
|
Nd
|
|
Chern et coll., 1980
|
399
|
609
|
0,22
|
0,04
|
|
Kaur et Singh, 1981
|
72
|
72
|
0,39
|
Nd
|
|
Brooks-Gunn et Warren, 1988
|
350
|
350 (63 étaient danseuses)
|
0,26
0,32
|
Non danseuses
Danseuses
|
|
Malina et coll., 1994
|
109
|
109
|
0,25
|
Nd
|
|
Graber et coll., 1995
|
75
|
75
|
0,23
|
Ajusté sur l'âge
|
|
Cameron et Nagdee, 1996
|
146
|
146
|
0,23
|
(0,16 après ajustement sur l'âge au rappel)
|
|
Nd : non disponible
|
En plus des études de corrélation qui renseignent sur la base génétique du trait observé, les études de jumeaux permettent de quantifier cette composante par rapport à celle non héritable. L'héritabilité (h
2) de l'âge de la ménarche varie de 0,44 à 0,95 suivant les études (Treloar et Martin, 1990
; Meyer et coll., 1991
; Kaprio et coll., 1995
; Loesch et coll., 1995
; Snieder et coll., 1998
; Kirk et coll., 2001
) (tableau 5.II
). Cette variabilité importante montre que les effets de l'environnement dans le déterminisme de l'âge de la ménarche varient en fonction des populations, de leur localisation géographique et pourraient également varier en fonction du temps. Les facteurs environnementaux impliqués dans cette variabilité restent à déterminer précisément.
Tableau 5.II Héritabilité de l'âge de la ménarche : corrélations selon des études de jumeaux monozygotes (MZ) et dizygotes (DZ) (d'après Towne et coll., 2005)
|
Références
|
Effectifs
|
Mesure de ressemblance entre jumeaux
|
| |
Nbre de MZ
|
Nbre de DZ
| |
| | | |
Intervalle moyen (mois)
|
| | | |
MZ
|
DZ
|
|
Tisserand-Perrier, 1953
|
46
|
39
|
2,2
|
8,2
|
|
Fischbein, 1977
|
28
|
48
|
3,5
|
8,5
|
|
Golden, 1981
|
290
|
217
|
7,4
|
14,2
|
|
Kaprio et coll., 1995
|
234
|
189
|
6,0
|
11,2
|
| | | |
Coefficient de corrélation (r)
|
| | | |
MZ
|
DZ
|
|
Gedda et Brenci, 1975
|
71
|
81
|
0,89
|
0,66
|
|
Fischbein, 1977
|
28
|
48
|
0,93
|
0,62
|
|
Golden, 1981
|
290
|
217
|
0,71
|
0,29
|
| | | |
Coefficient d'héritabilité h2 (± écart-type)
|
|
Van den Akker et coll., 1987 Londres
|
364 paires, jumeaux non spécifiés
|
0,72
|
Nd
|
|
Van den Akker et coll., 1987 Birmingham
|
98 paires, jumeaux non spécifiés
|
0,54
|
Nd
|
|
Treloar et Martin, 1990
|
1 177
|
711
|
0,61
|
0,68 (selon l'année de naissances)
|
|
Meyer et coll., 1991
|
1 233
|
751
|
0,71
|
Nd
|
|
Kaprio et coll., 1995
|
234
|
189
|
0,74
|
Nd
|
|
Loesch et coll., 1995
|
44
|
42
|
0,95
|
Nd
|
|
Snieder et coll., 1998
|
275
|
353
|
0,45
|
Nd
|
|
Kirk et coll., 2001
|
1 373
|
1 310
|
0,50
|
Nd
|
|
Eaves et coll., 2004
|
732
|
680
|
0,96
|
± 0,03
|
|
Mustanski et coll., 2004
|
647
|
1 244
|
0,94
|
Nd
|
|
Nd : non disponible
|
Analyse du déterminisme génétique de l'âge de la puberté : une méthodologie difficile
La plupart de ces études sont rétrospectives et basées sur le souvenir de l'âge des premières règles chez des femmes adultes. Le facteur de corrélation entre le souvenir et la réalité de l'âge de la ménarche varie entre 0,6 et 0,9 (Mustanski et coll., 2004
). Towne et coll. (2005
) ont utilisé les données de la cohorte « Fels » afin de diminuer l'incertitude de l'analyse rétrospective. L'étude Fels est une étude longitudinale, commencée en 1929 qui comprenait 200 familles en 2005 : 112 ont pu être étudiées, ce qui représente un échantillon de 371 femmes. Il ressort des résultats de Towne que le modèle le plus adapté est un modèle polygénique prenant en compte l'évolution séculaire de l'âge de la puberté. Pour ce modèle, l'héritabilité est de 0,49±0,13 ce qui indique que la moitié de la variabilité de l'âge de la ménarche dépend de facteurs génétiques.
Le manque de spécificité de la variable « ménarche » pour déterminer l'âge de la puberté est bien admis. La ménarche est le témoin d'un processus d'interactions hormonales complexes qui dépend de la production endogène des hormones stéroïdes sexuelles, de la sensibilité des tissus périphériques à ces hormones. Ce mécanisme physiologique dépend également de facteurs « perturbateurs » exogènes ayant une activité de type stéroïde. Cette « pollution » est également vraie pour l'appréciation du stade de développement des seins alors que l'obésité des pays développés est un autre facteur diminuant la spécificité de l'analyse (Parent et coll., 2003
). Certaines études ont contourné ce problème en assimilant la puberté à une variable dynamique définie par un score qui prend en compte plusieurs paramètres cliniques tels que le pic de croissance, l'apparition des poils pubiens, le développement des seins, les règles et les modifications de la peau ou le changement de la voix chez le garçon (Eaves et coll., 2004
; Mustanski et coll., 2004
). À partir d'une approche longitudinale chez 1 891 paires de jumeaux de 11 à 14 ans, un groupe finlandais rapporte une héritabilité proche de 1 déterminant l'âge du développement pubertaire défini selon le score ci-dessus. Ce groupe considère que la part de l'environnement devient alors négligeable par comparaison avec la ménarche. Cette approche est intéressante, en intégrant le pic de croissance pubertaire, elle évalue la maturation neuronale globale et pas seulement celle permettant la réactivation de l'axe gonadotrope. Ces résultats sont contradictoires avec l'évolution séculaire de l'âge de la puberté qui serait due à l'amélioration des conditions de vie et probablement en rapport avec les migrations d'un environnement rural vers un environnement urbain. L'analyse d'une population issue de la même région pendant un intervalle de temps court diminue le poids de la variation. Ces données sont confirmées par l'étude de Eaves et coll. (2004
) qui évalue l'héritabilité à 0,96.
Ces travaux montrent que la méthodologie est primordiale et que les résultats doivent être analysés en fonction de la question posée. Il est différent d'analyser l'âge de l'initiation de la puberté de l'âge de la ménarche bien que le deuxième dépende directement du premier. L'âge de la puberté est bien déterminé par des facteurs génétiques. Les facteurs de l'environnement sont plus ou moins importants suivant les espèces. L'exemple le plus connu étant le hamster sibérien dont la reproduction saisonnière dépend principalement de l'alternance photopériode courte/photopériode longue. La photopériode était probablement un élément important de la régulation de la reproduction chez les Inuits du Canada avant l'occidentalisation de leur mode de vie (Condon, 1991
).
Il semble maintenant admis que l'évolution séculaire de l'âge de la puberté a atteint un plancher, ce qui suggère que les facteurs de l'environnement participent maintenant a minima à la maturation normale de l'axe gonadotrope dans l'espèce humaine. Cependant, à fortes concentrations ou bien au cours d'une exposition chronique, ils pourraient perturber le fragile équilibre activation/inhibition. Il est fort probable que les effets de ces facteurs de l'environnement sur l'âge de la puberté dépendent directement du terrain génétique de chaque individu. Cette approche méthodologique mélangeant génétique et écologie, rassemblée sous la bannière « écogénétique » devient une nécessité en reproduction.
Gènes candidats au déterminisme génétique de l'âge de la puberté
Les modèles génétiques indiquent que l'âge de la puberté dépend de l'interaction en réseau de plusieurs gènes. Cette impression a été confirmée à partir de l'étude des maladies révélées par une anomalie de l'âge de l'initiation de la puberté. Il faut distinguer deux grands groupes : les anomalies de la commande neuroendocrinienne également appelées anomalies centrales, des anomalies gonadiques dites périphériques (Kalantaridou et Chrousos, 2002
; Gracia et Driscoll, 2003
). Ce point est fondamental. Dans le premier cas, il s'agit d'une anomalie de la maturation de l'axe gonadotrope alors que dans les formes périphériques, il s'agit d'une anomalie de la synthèse des hormones sexuelles ou de leurs effets sur les organes cibles. L'avance pubertaire ou bien l'absence de puberté peuvent être isolées ou bien syndromiques. Le phénotype peut être acquis, résulter d'une maladie chronique mais il peut également s'agir de maladies monogéniques. Dans les retards pubertaires d'origine génétique, plusieurs modes de transmission ont été décrits : lié au chromosome X, autosomique dominant ou récessif. Ces modèles de transmission sont en faveur d'un gène majeur. À l'inverse, le modèle de transmission de l'avance pubertaire par précocité de la maturation neuroendocrinienne (puberté précoce centrale) est clairement polygénique et multifactoriel (Parent et coll., 2003
). Les arguments en faveur d'un modèle monogénique avec transmission dominante pour la puberté précoce sont probablement dus à une surestimation du nombre de sujets atteints dans les familles étudiées (de Vries et coll., 2004). L'avance pubertaire centrale ne résulte probablement pas de l'activation isolée d'un gène mais plutôt d'une maturation neuroendocrinienne trop précoce dans son ensemble. Il existe, en revanche, plusieurs exemples de puberté précoce due à une sécrétion autonome et inappropriée d'hormones sexuelles par les gonades, dont certaines correspondent à des maladies monogéniques bien caractérisées (Huhtaniemi, 2002
; Kalantaridou et coll., 2002
).
Études d'association entre puberté précoce et gènes candidats
Deux approches expérimentales sont poursuivies pour comprendre la génétique des maladies de l'initiation de la puberté. Dans le cas de l'avance pubertaire, l'absence d'argument en faveur d'un gène majeur a incité la recherche des polymorphismes parmi des gènes candidats choisis selon des critères plus ou moins pertinents tels que les enzymes de la stéroïdogenèse. La ménarche précoce étant un facteur de risque connu du cancer du sein, plusieurs équipes ont testé des gènes candidats dans des études d'association en utilisant des polymorphismes de gènes des enzymes de la synthèse ou du catabolisme des stéroïdes sexuels. Les résultats sont discordants en fonction du trait étudié. Une association est décrite entre un variant du gène
CYP3A41B qui intervient dans le catabolisme de la testostérone et le développement mammaire à un âge donné (9 ans) (Kadlubar et coll., 2003
). Un autre variant du gène
CYP3A4 semble associé à la puberté précoce dans une étude chinoise (Xin et coll., 2005
) alors qu'aucune association n'a été retrouvée par Lai et coll. (2001
). Il faut souligner que la définition du phénotype étudié n'est pas identique dans toutes les études, ce qui gène la comparaison des résultats.
Génétique du retard pubertaire par déficit gonadotrope
Depuis le début des années 2000, la séquence complète du génome humain a fortement aidé à la compréhension des formes familiales de déficit gonadotrope par des approches de gènes candidats ou de cartographie du génome. Certains gènes participent au développement fœtal des neurones à GnRH (
Gonadotropin Releasing Hormone) tels que le gène
KAL1 (Franco et coll., 1991
; Legouis et coll., 1991
) ou le gène
FGFR1 qui code le récepteur 1 du FGF (
Fibroblast Growth Factor) (Dode et coll., 2003
). Les gènes décrits dans la régulation supra-hypothalamique postnatale sont multiples et pour l'instant aucune anomalie génétique n'a été décrite dans l'espèce humaine. Récemment, ces approches de génétique ont permis d'impliquer, dans la régulation hypothalamique de l'axe gonadotrope, la leptine (Strobel et coll., 1998
) et son récepteur (Clement et coll., 1998
),
DAX-1 (Zanaria et coll., 1994
),
SF1 (Achermann et coll., 1999
),
PC-1 (O'Rahilly et coll., 1995),
PROP1 (Wu et coll., 1998
) et récemment le récepteur
GPR54 (de Roux et coll., 2003 ; Seminara et coll., 2003
). Le rôle des gènes hypophysaires tels que ceux codant le récepteur de la GnRH (de Roux et coll., 1997), la LH (Phillip et coll., 1998
) ou la FSH (Matthews et coll., 1993
) est bien connu depuis plusieurs années bien que la description des anomalies génétiques au sein de ces gènes soit récente.
Tous les phénotypes décrits jusqu'à ce jour résultent de mutations de type « perte de fonction » entraînant une absence de puberté. Les phénotypes sont variés et dépendent en partie des autres fonctions biologiques des gènes concernés. Les mutations « perte de fonction » des gènes KAL1 et FGFR1 sont décrites dans le syndrome de Kallmann qui associe anosmie et déficit gonadotrope. Cette association est actuellement expliquée par le défaut de migration des neurones GnRH dû à l'agénésie du bulbe olfactif. La très grande variabilité de l'expression phénotypique des mutations « perte de fonction » du FGFR1 suggère que la relation de cause à effet entre agénésie des bulbes et défaut de migration des neurones n'est pas certaine. Les mutations « perte de fonction » de la leptine et de son récepteur associent une obésité majeure au déficit gonadotrope. Cette association est expliquée par le rôle neuroendocrinien de la leptine dans la régulation de la faim et de l'axe gonadotrope. Dans les anomalies de DAX-1, le déficit gonadotrope est associé à une insuffisance surrénale.
La description des mutations « perte de fonction » du récepteur GPR54 dans le déficit gonadotrope isolé a été une avancée majeure dans la physiologie de l'axe gonadotrope et de l'initiation de la puberté. La puissance de l'analyse des maladies génétiques monofactorielles pour mieux comprendre la physiologie et la génétique d'un trait polygénique, voire multifactoriel, est bien illustrée par cet exemple. Ce récepteur n'était pas candidat car il était connu pour son activité inhibitrice de métastases (Karges et de Roux, 2005
; Dungan et coll., 2006
). Une approche de cartographie du génome a permis de le considérer comme candidat (de Roux et coll., 2003 ; Seminara et coll., 2003
). À partir de ce travail, plusieurs groupes ont démontré que les ligands de
GPR54, les peptides Kiss-1 (Kp), étaient des puissants sécrétagogues des hormones GnRH, LH et FSH. L'augmentation de l'expression hypothalamique du gène
Kiss-1 à la puberté est un autre argument soulignant son rôle dans les mécanismes d'initiation de la puberté. La présence d'un hypogonadisme à la naissance chez les patients porteurs d'une mutation inactivatrice de
GPR54 et la persistance de l'hypogonadisme à l'âge adulte ont montré que le système Kiss-1/GPR54 est un modulateur de l'axe gonadotrope de la vie fœtale à la vie adulte et pas seulement un initiateur de la puberté.
La description des mutations inactivatrices du récepteur de la GnRH a permis de mieux comprendre plusieurs phénotypes dont les eunuques fertiles (Karges et coll., 2005
). Le phénotype de ces patients comprend un hypogonadisme par déficit gonadotrope associé à une spermatogenèse normale. Les valeurs de LH et FSH sont dans les limites de la normale alors que la testostérone est basse. Les mutations inactivatrices du récepteur de la GnRH caractérisées chez ces patients inhibent partiellement la fonction du récepteur
in vitro. De plus, une expressivité variable du phénotype a été décrite au sein de plusieurs familles, ce qui suggère que d'autres facteurs génétiques ou environnementaux peuvent moduler l'hypogonadisme dû à une mutation du récepteur de la GnRH (de Roux et coll., 1999). Ce point confirme le caractère multifactoriel de la régulation de l'axe gonadotrope même dans une situation de déficit pathologique profond.
Tous ces travaux sur la génétique du retard pubertaire par défaut de la régulation neuroendocrinienne de l'axe gonadotrope ont apporté des éléments nouveaux en décrivant l'implication de nouveaux gènes tels que KAL1, FGFR1, DAX1, SF1, PROP1, le gène codant le récepteur de la leptine, GPR54, ou en caractérisant des anomalies dans des gènes connus tels que GnRHR, LH et FSH. Il est possible que la variabilité de l'âge de la puberté dans la population dépende en partie de polymorphismes au sein des séquences codantes ou régulatrices de ces gènes ou de leurs partenaires.
Puberté avancée et survenue de pathologies durant la vie adulte : existe-t-il un lien génétique ?
La puberté précoce, et notamment la ménarche précoce, sont des facteurs de risque connus du cancer du sein. Plusieurs hypothèses pourraient expliquer cette observation. Une puberté précoce augmente le temps d'exposition aux œstrogènes ou à des molécules
estrogènes like et ceci à un âge plus précoce. Elle indique également une activation prématurée de l'axe gonadotrope dont l'origine génétique pourrait être en rapport avec la prédisposition génétique à développer un cancer dépendant des hormones sexuelles. Pour tenter de répondre à cette question, Hamilton et Mack (2003
) ont comparé le risque de cancer du sein chez des jumelles monozygotes en fonction de l'âge de la puberté. Leurs résultats montrent que le cancer du sein survient plus tôt chez la jumelle monozygote possédant un risque génétique élevé de cancer du sein et ayant eu le développement pubertaire le plus précoce. Ce résultat n'est pas observé chez les jumelles dizygotes. Ce risque ne semble pas être modulé par les expositions ultérieures aux hormones sexuelles, ce qui suggère que cet effet est dû à l'exposition hormonale au moment de la puberté. Dans la même IDée, Remsberg et coll. (2005
) ont rapporté à partir de l'étude Fels un risque plus important de développement de facteurs de risque cardiovasculaire (glycémie, insulinémie, pression artérielle) chez les adolescentes ayant eu des règles précoces. La survenue précoce de la ménarche est associée à une élévation de la pression artérielle et une plus forte proportion d'intolérance au glucose.
En conclusion,
la génétique de l'âge de la puberté est complexe. Il existe des gènes majeurs dont le déficit est responsable de retard pubertaire majeur avec infertilité. Une part non négligeable du déterminisme de l'âge de l'initiation de la puberté dépend certainement de polymorphismes au sein de ces gènes majeurs. Les facteurs de l'environnement perturbent facilement le fragile équilibre entre l'activation et l'inhibition de l'axe gonadotrope. Au fur et à mesure que la puissance de l'approche génétique augmente, il faut améliorer celle de l'analyse phénotypique. Ce point est certainement actuellement l'inconvénient majeur de ces études. Il devient indispensable de définir de nouveaux marqueurs cliniques mais surtout biologiques des phases précoces de la puberté. Une meilleure compréhension du lien entre initiation de la puberté, environnement et survenue de pathologies dépendant directement des hormones sexuelles passe obligatoirement par la mise en place d'études épidémiologiques longitudinales à grande échelle comprenant une approche clinique, biologique et génétique.
Bibliographie
[1] achermann jc,
ito m,
hindmarsh pc,
jameson jl. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans.
Nat Genet. 1999;
22:125
-126

[2] bolk l. Untersuchungen über die Menarche bei der niederlandischen Bevolkerung.
Z Geburtshilfe Gynakol. 1926;
89:364
-380
[3] brooks-gunn j,
warren mp. Mother-daughter differences in menarcheal age in adolescent girls attending national dance company schools and non-dancers.
Ann Hum Biol. 1988;
15:35
-44
[4] cameron n,
nagdee i. Menarcheal age in two generations of South African Indians.
Ann Hum Biol. 1996;
23:113
-119
[5] chern mm,
gatewood lc,
anderson ve. The inheritance of mensual traits.
In: dan aj, graham ea, beecher cp, editors.
The menstrual cycle: a synthesis of interdisciplinary research..
New York:Springer;
1980.
p. 123
-130
[6] clement k,
vaisse c,
lahlou n,
cabrol s,
pelloux v. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction.
Nature. 1998;
392:398
-401
[7] condon rg. Birth seasonality, photoperiod, and social change in the central Canadian Arctic.
Hum Ecol. 1991;
19:287
-321
[8] damon a,
damon st,
reed rb,
valadian i. Age at menarche of mothers and daughters, with a note on accuracy of recall.
Hum Biol. 1969;
41:161
-175
[9] de roux n,
young j,
misrahi m,
genet r,
chanson p. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor.
N Engl J Med. 1997;
337:1597
-1602
[10] de roux n,
young j,
brailly-tabard s,
misrahi m,
milgrom e,
schaison g. The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred.
J Clin Endocrinol Metab. 1999;
84:567
-572
[11] de roux n,
genin e,
carel jc,
matsuda f,
chaussain jl,
milgrom e. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54.
Proc Natl Acad Sci USA. 2003;
100:10972
-10976
[12] de vries l,
kauschansky a,
shohat m,
phillip m. Familial central precocious puberty suggests autosomal dominant inheritance.
J Clin Endocrinol Metab. 2004;
89:1794
-1800
[13] dode c,
levilliers j,
dupont jm,
de paepe a,
le du n. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome.
Nat Genet. 2003;
33:463
-465
[14] dungan hm,
clifton dk,
steiner ra. Minireview: kisspeptin neurons as central processors in the regulation of gonadotropin-releasing hormone secretion.
Endocrinology. 2006;
147:1154
-1158
[15] eaves l,
silberg j,
foley d,
bulik c,
maes h. Genetic and environmental influences on the relative timing of pubertal change.
Twin Res. 2004;
7:471
-481
[16] ebling fj. The neuroendocrine timing of puberty.
Reproduction. 2005;
129:675
-683
[17] fischbein s. Onset of puberty in MX and DZ twins.
Acta Genet Med Gemellol (Roma). 1977;
26:151
-158
[18] franco b,
guioli s,
pragliola a,
incerti b,
bardoni b. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules.
Nature. 1991;
353:529
-536
[19] gedda l,
brenci g. Twins as a natural test of chronogenetics.
Acta Genet Med Gemellol (Roma). 1975;
24:15
-30
[20] golden wl. Reproductive histories in a Norwegian twin population: evaluation of the maternal effect in early spontaneous abortion.
Acta Genet Med Gemellol (Roma). 1981;
30:91
-165
[21] graber ja,
brooks-gunn j,
warren mp. The antecedents of menarcheal age: heredity, family environment, and stressful life events.
Child Dev. 1995;
66:346
-359
[22] gracia cr,
driscoll da. Molecular basis of pubertal abnormalities.
Obstet Gynecol Clin North Am. 2003;
30:261
-277
[23] hamilton as,
mack tm. Puberty and genetic susceptibility to breast cancer in a case-control study in twins.
N Engl J Med. 2003;
348:2313
-2322
[24] huhtaniemi it. LH and FSH receptor mutations and their effects on puberty.
Horm Res. 2002;
57Suppl 2:35
-38
[25] israel s. Onset of menstruation in Indian women.
J Obstet Gynaecol Br Emp. 1959;
66:311
-316
[26] kadlubar ff,
berkowitz gs,
delongchamp rr,
wang c,
green bl. The CYP3A4*1B variant is related to the onset of puberty, a known risk factor for the development of breast cancer.
Cancer Epidemiol Biomarkers Prev. 2003;
12:327
-331
[27] kalantaridou sn,
chrousos gp. Clinical review 148: Monogenic disorders of puberty.
J Clin Endocrinol Metab. 2002;
87:2481
-2494
[28] kantero r-l,
widholm o. Correlations of menstrual traits between adolescent girls and their mothers.
Acta Obstet Gynecol Scand. 1971;
14:30
-36
[29] kaprio j,
rimpela a,
winter t,
viken rj,
rimpela m,
rose rj. Common genetic influences on BMI and age at menarche.
Hum Biol. 1995;
67:739
-753
[30] karges b,
de roux n. Molecular genetics of isolated hypogonadotropic hypogonadism and Kallmann syndrome.
Endocr Dev. 2005;
8:67
-80
[31] kaur dp,
singh r. Parent-adult offspring correlations and heritability of body measurements in a rural Indian population.
Ann Hum Biol. 1981;
8:333
-339
[32] kirk km,
blomberg sp,
duffy dl,
heath ac,
owens ip,
martin ng. Natural selection and quantitative genetics of life-history traits in Western women: a twin study.
Evolution Int J Org Evolution. 2001;
55:423
-435
[33] lai j,
vesprini d,
chu w,
jernstrom h,
narod sa. CYP gene polymorphisms and early menarche.
Mol Genet Metab. 2001;
74:449
-457
[34] legouis r,
hardelin jp,
levilliers j,
claverie jm,
compain s. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules.
Cell. 1991;
67:423
-435
[35] loesch dz,
huggins r,
rogucka e,
hoang nh,
hopper jl. Genetic correlates of menarcheal age: a multivariate twin study.
Ann Hum Biol. 1995;
22:470
-490
[36] malina rm,
ryan rc,
bonci cm. Age at menarche in athletes and their mothers and sisters.
Ann Hum Biol. 1994;
21:417
-422
[37] manasco pk,
umbach dm,
muly sm,
godwin dc,
negro-vilar a. Ontogeny of gonadotropin, testosterone, and inhibin secretion in normal boys through puberty based on overnight serial sampling.
J Clin Endocrinol Metab. 1995;
80:2046
-2052
[38] manasco pk,
umbach dm,
muly sm,
godwin dc,
negro-vilar a. Ontogeny of gonadotrophin and inhibin secretion in normal girls through puberty based on overnight serial sampling and a comparison with normal boys.
Hum Reprod. 1997;
12:2108
-2114
[39] matthews ch,
borgato s,
beck-peccoz p,
adams m,
tone y. Primary amenorrhoea and infertility due to a mutation in the beta-subunit of follicle-stimulating hormone.
Nat Genet. 1993;
5:83
-86
[40] meyer jm,
eaves lj,
heath ac,
martin ng. Estimating genetic influences on the age-at-menarche: a survival analysis approach.
Am J Med Genet. 1991;
39:148
-154
[41] mustanski bs,
viken rj,
kaprio j,
pulkkinen l,
rose rj. Genetic and environmental influences on pubertal development: longitudinal data from Finnish twins at ages 11 and 14.
Dev Psychol. 2004;
40:1188
-1198
[42] o’rahilly s,
gray h,
humphreys pj,
krook a,
polonsky ks. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function.
N Engl J Med. 1995;
333:1386
-1390
[43] parent as,
teilmann g,
juul a,
skakkebaek ne,
toppari j,
bourguignon jp. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration.
Endocr Rev. 2003;
24:668
-693
[44] phillip m,
arbelle je,
segev y,
parvari r. Male hypogonadism due to a mutation in the gene for the beta-subunit of follicle-stimulating hormone.
N Engl J Med. 1998;
338:1729
-1732
[45] remsberg ke,
demerath ew,
schubert cm,
chumlea wc,
sun ss,
siervogel rm. Early menarche and the development of cardiovascular disease risk factors in adolescent girls: the Fels Longitudinal Study.
J Clin Endocrinol Metab. 2005;
90:2718
-2724
[46] rogan wj,
ragan nb. Evidence of effects of environmental chemicals on the endocrine system in children.
Pediatrics. 2003;
112:247
-252
[47] seminara sb,
messager s,
chatzidaki ee,
thresher rr,
acierno js jr. The GPR54 gene as a regulator of puberty.
N Engl J Med. 2003;
349:1614
-1627
[48] snieder h,
macgregor aj,
spector td. Genes control the cessation of a woman’s reproductive life: a twin study of hysterectomy and age at menopause.
J Clin Endocrinol Metab. 1998;
83:1875
-1880
[49] strobel a,
issad t,
camoin l,
ozata m,
strosberg ad. A leptin missense mutation associated with hypogonadism and morbid obesity.
Nat Genet. 1998;
18:213
-215
[50] tisserand-perrier m. Étude comparative de certains processus de croissance chez les jumeaux.
J Genet Hum. 1953;
2:87
-102
[51] towne b,
czerwinski sa,
demerath ew,
blangero j,
roche af,
siervogel rm. Heritability of age at menarche in girls from the Fels Longitudinal Study.
Am J Phys Anthropol. 2005;
128:210
-219
[52] treloar sa,
martin ng. Age at menarche as a fitness trait: nonadditive genetic variance detected in a large twin sample.
Am J Hum Genet. 1990;
47:137
-148
[53] van den akker oba,
stein gs,
neale mc,
murray rm. Genetic and environmental variation in menstrual cycle: histories of two British twin samples.
Acta Genet Med Gemellol (Roma). 1987;
36:541
-548
[54] wu w,
cogan jd,
pfaffle rw,
dasen js,
frisch h. Mutations in PROP1 cause familial combined pituitary hormone deficiency.
Nat Genet. 1998;
18:147
-149
[55] wyshak g,
frisch re. Evidence for a secular trend in age of menarche.
N Engl J Med. 1982;
306:1033
-1035
[56] xin x,
luan x,
xiao j,
wei d,
wang j,
lu d,
yang s. Association study of four activity SNPs of CYP3A4 with the precocious puberty in Chinese girls.
Neurosci Lett. 2005;
381:284
-288
[57] zacharias l,
rand wm,
wurtman rj. A prospective study of sexual development and growth in American girls: the statistics of menarche.
Obstet Gynecol Surv. 1976;
31:325
-337
[58] zanaria e,
muscatelli f,
bardoni b,
strom tm,
guioli s. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita.
Nature. 1994;
372:635
-641