Réduction des dommages associés à la consommation d’alcool
I. Consommations d’alcool : les risques, les dommages et leur environnement
2021
5-
Impact des données
en épigénétique
Notre but dans ce chapitre est d’évaluer l’impact des découvertes les plus
récentes en épigénétique et de la consommation d’alcool sur les possibilités
diagnostiques et thérapeutiques qu’elles ouvrent et leur impact en termes de
prévention et de réduction des risques. Nous nous limiterons aux études qui
concernent les déficits du développement et de l’intégrité du cerveau,
principalement en réponse à l’exposition prénatale à l’alcool. En effet, les
problématiques soulevées par ce domaine couvrent et illustrent parfaitement
les enjeux de l’épigénétique pour la prévention et réduction des risques
liés aux consommations à risque
1
Pour une meilleure compréhension de ce chapitre, voici
la liste des abréviations : ARNm, Acide ribonucléique messager
codant une (des) protéine(s) ; EPA, Exposition prénatale à
l’alcool ; cellules neurales (« nerveuses », neurones, astrocytes,
oligodendrocytes). Acteurs épigénétiques touchant les résidus
d’acides aminés lysine d’histones ou de protéines non-histones :
HAT/KAT, acétyl-lysine/histone-transférase ; HDAC/KDAC,
lysine/histone-désacétylase ; HTM/KMT,
méthyl-lysine/histone-transférase ; HDM/KDM,
lysine/histone-déméthylase. Acteurs épigénétiques liés à la
méthylation de l’ADN sur des cytosines : DNMT,
DNA-5mC-méthyl-transférase ; TET, 5mC-hydroxy-méthylase impliquée
dans la déméthylation de l’ADN.
.
Développement du cerveau et impact épigénétique de
l’alcoolisation fœtale
Le développement du cerveau et son fonctionnement à l’âge adulte sont
étroitement contrôlés par des mécanismes épigénétiques (Bird,
2007

).
L’importance de ce contrôle épigénétique est soulignée par le grand
nombre de maladies neuro-développementales et neuropsychiatriques
associées à des mutations ou variants de gènes codant des acteurs
épigénétiques (syndrome de Rett, syndrome de Rubinstein-Taybi, autisme,
schizophrénie...) (Gräff et coll., 2011
; LaSalle et coll.,
2013
;
Bourgeron, 2015
;
Liu et coll., 2016a
).
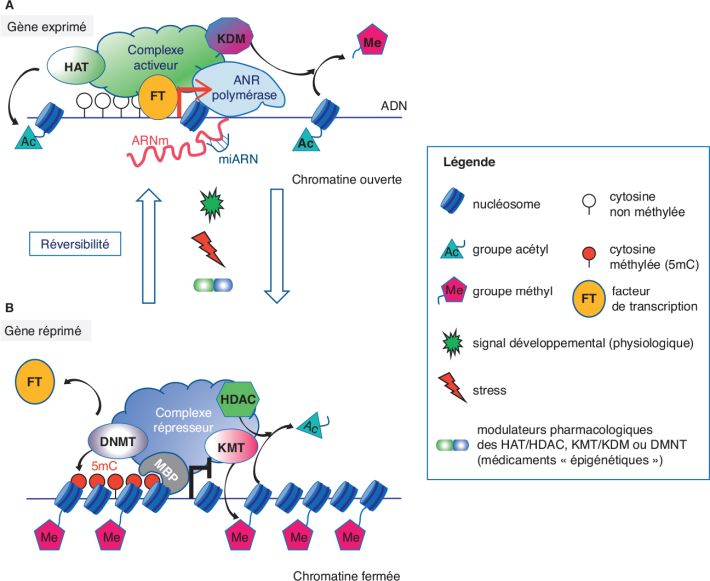
Mécanismes épigénétiques
Les mécanismes épigénétiques contrôlent l’expression des gènes, sans
modifier la séquence de l’ADN (acide désoxyribonucléique, support de
l’information génétique et de sa transmission au cours des
générations) elle-même, mais en contrôlant l’accessibilité de
l’information génétique contenue dans l’ADN aux machineries
cellulaires qui vont lire et transcrire cette information (mécanisme
dit de « transcription ») et permettre ainsi, ou non, à la cellule
« d’exprimer » l’information contenue dans tel ou tel gène, par la
synthèse de protéines spécifiques ou d’ARN dits non-codants (ne
codant pas de protéines) (figure 5.1
). L’ADN est une molécule longue d’environ
deux mètres, dans une cellule de mammifère, qui est compactée dans
un noyau de quelques microns, ce qui compromet, en effet,
l’accessibilité à son information génétique, mais procure aussi un
niveau crucial de régulation de l’expression des gènes.
La compaction de l’ADN dans le noyau est assurée par son enroulement
autour d’un complexe protéique, composés d’histones, ce complexe
constituant le nucléosome, c’est-à-dire l’unité de base qui
constitue la chromatine (figure 5.1
). Cette unité de base, ainsi que des ordres supérieurs
d’enroulement, limitent l’accessibilité à l’information génétique
qui est contenue dans cette portion de l’ADN (Dulac,
2010
;
Schang et coll., 2018
). Les mécanismes épigénétiques agissent
sur cette compaction et donc « l’ouverture » ou la « fermeture » de
la chromatine, par des modifications chimiques (modifications
post-traductionnelles) (figure 5.1
). Ces « décorations » chimiques constituent le « code histone »
ou plutôt un « langage histone ». Elles sont héritables d’une
division cellulaire à l’autre, ou acquises
de novo, et sont
très diverses : acétylation, méthylation, phosphorylation... pour ne
citer que les plus étudiées. Elles induisent soit une stabilisation,
soit au contraire une déstabilisation des nucléosomes, en attirant
de gros complexes, répresseurs ou activateurs, respectivement. Ces
complexes contiennent des acteurs épigénétiques
(« scribes/écrivains »), responsables du dépôt de ces modifications
chimiques (marques épigénétiques) qui, lues par des « lecteurs »,
provoquent le dépôt de nucléosomes additionnels grâce à
l’intervention de « remodeleurs » de la chromatine, et donc une
augmentation de l’enroulement – ou bien leur éviction, ce qui induit
le déroulement de l’ADN. On aboutit à une conformation dite fermée
ou ouverte de la chromatine, respectivement. La modification
épigénétique la plus étudiée est la méthylation de l’ADN lui-même.
Elle s’effectue par l’ajout de groupement méthyl (-CH3) sur des
résidus cytosines (C) – une des quatre bases, qui forment l’ADN –
les autres étant l’adénine (A), cytosine (C), la guanine (G) et la
thymine (T), dont l’ordre constitue la séquence de l’ADN, support de
l’information génétique. D’une manière très schématique, la
méthylation de l’ADN dans un contexte « CpG » (cytosines, suivies
d’une guanine) situées au cœur des régions régulatrices de
l’expression des gènes, a globalement pour effet de condenser la
chromatine, aboutissant à une conformation fermée de la chromatine
(figure 5.1
). La distribution
des CpG dans le génome fait apparaître l’existence d’îlots de CpG
(regroupements de CpG), situés à proximité des promoteurs de gènes,
et dont la méthylation induit une répression de l’expression des
gènes. Les gros complexes contiennent aussi de (longs) ARN
non-codants qui, ne codent pas des protéines, mais, qui, enveloppant
une région, peuvent en particulier agir comme pièges ou « éponges »,
et maintenir les acteurs épigénétiques à proximité du site à
remodeler (Briggs et coll., 2015
). Enfin, le contrôle épigénétique de
l’expression d’un gène implique aussi les petits ARN non-codants,
dont les micro-ARN (miARN), qui, grâce à des homologies de séquences
avec des ARN messagers (ARNm
2
Acide ribonucléique messager codant une (des)
protéine(s).
) régulent leur niveau de traduction en protéines.
Un miARN peut réguler la traduction d’ARNm de plusieurs gènes et
l’expression d’un gène est régulée par plusieurs miARN (Yi et Fuchs,
2011
).
Ces mécanismes épigénétiques sont à l’œuvre tout au long des étapes
qui régissent la construction du cerveau, son intégrité et ses
fonctions : la prolifération des progéniteurs neuraux, la migration
des jeunes neurones, la formation des axones et des dendrites, celle
des synapses et, enfin, la plasticité neuronale. L’activité
neuronale elle-même est capable de modifier l’épigénome,
c’est-à-dire les caractéristiques des marques épigénétiques et la
conformation de la chromatine à l’échelle du génome (Su et coll.,
2017
). De
plus, une dynamique très particulière de formes de méthylation dans
des contextes non-CpG a été identifiée dans le cerveau humain et de
souris, après la naissance dans le cortex préfrontal. Absente du
cerveau fœtal, elle est caractérisée par une augmentation brutale
pendant la petite enfance, et atteint son maximum à la fin de
l’adolescence. Cette forme de méthylation est très proéminente dans
les neurones en particulier (Lister et coll.,
2013
; He
et Ecker, 2015
; Schultz et coll., 2015
). Chez la souris, elle coïncide avec une
augmentation de l’ADN méthyl-transférase DNMT3A qui méthyle
de
novo les cytosines. Cette période correspond chez l’Homme et
la souris à une augmentation de la densité de synapses durant
l’enfance, puis avec l’élagage synaptique durant l’adolescence, ce
qui souligne le rôle proéminent de la méthylation de l’ADN dans le
développement et la fonction cérébrale. En outre, ces résultats
procurent un nouveau cadre de pensée pour la compréhension du rôle
de l’épigénome et de ses altérations dans le cerveau normal et
pathologique. De façon notable, la méthylation des cytosines dans un
contexte non-CpG n’est pas lue par la cellule selon les mêmes
grilles d’interprétation que la méthylation CpG, d’une part, avec
des impacts opposés sur l’expression des gènes, dans les cellules
souches et le cerveau mature.
Exposition prénatale à l’alcool et impacts épigénétiques
sur le cerveau en développement
La dérégulation de ces mécanismes épigénétiques affecte profondément le
développement du cerveau et son intégrité – que ce soit en raison de
mutations présentes dans des acteurs épigénétiques ou d’expositions à
des agressions environnementales au stade fœtal, périnatal ou adulte.
Des agressions très variées sont capables d’affecter le développement du
cerveau, et d’augmenter le risque de développer des maladies
neuropsychiatriques, selon le concept «
DOhaD » (
developmental
origins of health and disease) : l’exposition de la mère ou du
jeune enfant à la maltraitance (violences physiques et/ou psychologiques
et abus sexuels), la (mal)nutrition et l’exposition à des toxiques, dont
l’alcool, ou à des polluants (Thompson et coll.,
2009
; Ishii
et Hashimoto-Torii, 2015
; Guintivano et Kaminsky,
2016
; Schang
et coll., 2018
).
L’exposition prénatale à l’alcool (EPA) est considérée comme une cause
majeure d’anomalies neurodéveloppementales (Popova et coll.,
2012
). Elle
endommage l’ensemble des processus neuro-développementaux cités
ci-dessus, et donc tous les stades (Guerri et coll.,
2009
) :
prolifération et différenciation déficiente des progéniteurs neuraux,
migration neuronale anormale, mort neuronale, synaptogénèse compromise,
neurotransmission et plasticité neuronale perturbées. Outre provoquer
des avortements spontanés et des morts subites inexpliquées du
nourrisson, ces défauts contribuent à un large spectre de déficits dont
la sévérité est variable et qui, sur la base du travail pionnier de
Lemoine et coll. (1968
) et Jones et Smith
(1973
) est
répertorié sous le nom de « troubles causés par l’alcoolisation fœtale »
(ou TCAF ; FADS,
Fetal Alcohol Spectrum disorders (Sowell et
coll., 2001
)
(Streissguth et O’Malley, 2000
).
Ce spectre s’étend de défauts structurels et macroscopiques de diverses
régions cérébrales à des incapacités neurocomportementales plus
subtiles, avec parfois des traits autistiques (Mattson et coll.,
2011
;
Varadinova et Boyadjieva, 2015
). Dans le cas où des anomalies structurales
du cerveau sont décelées en imagerie cérébrale, on parle de Syndrome
d’alcoolisation fœtale (SAF). Les dysfonctionnements primaires du
cerveau affectent plus particulièrement le comportement et la cognition
et sont référencés dans l’outil de diagnostic mnémotechnique « ALARM »
Adaptive functioning (aptitudes sociales, maturité
émotionnelle, concepts de temps et d’argent, et compréhension),
Language/Learning,
Attention (impulsivité,
hyperactivité et difficultés d’attention, taux élevé de troubles de
l’attention/hyperactivité TDAH,
Reasoning (raisonnement et
fonctions exécutives),
Memory (mémoire verbale et non verbale)
(Inserm, 2001
;
Clarke et Gibbard, 2003
; Mattson et coll.,
2011
). Des
incapacités secondaires surviennent au cours de la vie de l’individu,
dont des maladies neuropsychiatriques avec, au premier plan, les
troubles de l’anxiété, la dépression majeure et la vulnérabilité aux
addictions (revues : Kodituwakku, 2007
; O’Connor et Paley,
2009
).
La difficulté inhérente au diagnostic d’un ensemble de troubles aussi
complexes et à la sévérité aussi variable que le TCAF, d’un individu à
l’autre, explique que les enfants sont souvent diagnostiqués
tardivement, généralement au moment de la scolarisation. La détection
des femmes enceintes à risque pour leur consommation à risque d’alcool
est également compliquée et reste très restreinte, et celle de leur père
l’est encore plus. La difficulté du diagnostic est encore accrue lorsque
qu’aucune donnée sur l’historique d’exposition n’est connue (dans les
cas d’adoption, par exemple). La nécessité d’identifier des biomarqueurs
d’EPA et possiblement des molécules à effet thérapeutique est donc
particulièrement aigüe en ce qui concerne le TCAF dont l’incidence est
manifestement sous-estimée (cf. chapitre « Mortalité, faibles
consommations et effets biologiques »). Les perturbations épigénétiques,
en raison de leur persistance et leur réversibilité potentielle sous
l’effet de traitements pharmacologiques ont ouvert une voie prometteuse
à cet égard.
EPA, TCAF et perturbations
épigénétiques
L’étude des modèles cellulaires, mais surtout animaux, a été déterminante
dans la compréhension des effets de l’EPA sur l’épigénome, d’une part,
parce que la taille des cohortes d’enfants ou de collections
d’échantillons de fœtus TCAF restent très limitées, et, d’autre part,
parce qu’ils ont permis « d’isoler » la composante « EPA » de facteurs
confondants (problèmes économiques et éducationnels, abus sexuels et
maltraitance, dépression chez la mère, addiction à d’autres substances
psychotropes...). Les études ont principalement impliqué des modèles de
rongeurs, mais pas uniquement (par exemple, des embryons de poulet et
des primates non-humains). Nous privilégierons les études portant sur
les mammifères.
Une des difficultés majeures rencontrée dans le domaine est la diversité
des modèles cellulaires et animaux, et des protocoles d’EPA : les doses
d’éthanol ; l’administration chronique, aiguë, ou intermittente ; la
fenêtre de développement du cerveau ; la durée de la consommation ou de
l’administration ; le laps de temps écoulé entre l’administration
d’alcool et les observations moléculaires, neuro-morphologiques ou
comportementales, le mode d’administration (gavage, injection
intra-péritonéale, voie orale (liquide ou semi-liquide). Même si l’on se
limite au modèle murin, le fond génétique varie considérablement d’une
souche de souris à l’autre ce qui là aussi peut conduire à des résultats
différents vis-à-vis des effets de l’alcool (Xu et coll.,
2019
). De
nombreuses études s’intéressent au cerveau entier, ce qui introduit un
biais parce que différentes régions du cerveau ne présentent
physiologiquement ni les mêmes profils de méthylation, ni la même
proportion de types cellulaires neuraux, puisque les profils de
méthylation sont très dépendants du type cellulaire (Lussier et coll.,
2018a
). D’une
manière générale, il n’est donc pas possible de définir une liste des
régions du génome différentiellement marquées épigénétiquement par
l’alcool. Tout au plus peut-on dégager des tendances et pointer vers de
futurs biomarqueurs potentiels.
Disponibilité des métabolites nécessaires au
fonctionnement
des acteurs épigénétiques
L’EPA engendre des bouleversements dans la disponibilité et la
synthèse de molécules clés du métabolisme cellulaire – en
particulier le métabolisme du carbone (
one-carbon) – qui ont
un retentissement sur l’activité des enzymes impliquées dans le
dépôt de marques épigénétiques. Inversement, ces études ont observé
une amélioration des endophénotypes du TCAF, par des traitements de
complémentation dans la nourriture par des métabolites. C’est le cas
du folate dont le transfert de la mère à l’embryon est altéré par
l’EPA (Hutson et coll., 2012
) et qui est un donneur de groupe méthyl,
indispensable à l’établissement et au maintien de la méthylation de
l’ADN ou des histones, et dont la carence est donc susceptible
d’engendrer des anomalies de l’épigénome). C’est aussi le cas de la
disponibilité en S-adénosyl-méthionine qui est aussi un donneur de
méthyl et dont les niveaux sont diminués, en raison du stress
oxydatif engendré par l’éthanol. L’apport en choline, qui est un
précurseur de S-adénosyl-homocytéine et de S-adénosyl-méthionine,
peut donc modifier aussi les marques épigénétiques de méthylation.
Enfin, le stress oxydatif engendré par l’exposition à l’éthanol
résulte aussi en une augmentation d’acétyl-CoA qui est un précurseur
de groupement acétyl- et utilisé par les HAT pour acétyler les
histones (Kleiber et coll., 2014
; Chater-Diehl et coll.,
2017
).
Altération globale des modifications des histones et
de la méthylation de l’ADN
De nombreuses études sur des modèles cellulaires, ou des coupes de
tissus de modèles animaux ou de fœtus ou jeunes enfants, morts
rapidement après la naissance, ont tenté une approche globale de la
caractérisation de modifications des signatures épigénétiques. Ces
études reposent sur la détection à l’aide d’anticorps de la
méthylation de l’ADN (voir pour un exemple récent portant sur des
échantillons de fœtus et de nouveau-nés TCAF
post mortem
(Jarmasz et coll., 2019
) ou de modifications post-traductionnelles
des histones (Chater-Diehl et coll.,
2017
).
D’autres encore évaluent globalement la méthylation de l’ADN et
l’activité de certains acteurs responsables du dépôt de marques
d’histones. Les résultats sont très variables selon les systèmes et
leur interprétation très limitée car ces données sont difficiles à
relier à l’expression génique ou au phénotype. Cependant, elles
suggèrent que l’EPA induit des perturbations globales de
disponibilité et d’activité d’acteurs épigénétiques (DMT, HAT/KAT
[acétyl-lysine/histone-transférase], HDAC/KDAC
[lysine/histone-désacétylase], KMT ou KDM) (Chater-Diehl et coll.,
2017
;
Veazey et coll., 2017
).
Impact de l’EPA sur les modifications
post-traductionnelles d’histones
Une revue de l’ensemble de ces modifications a été établie par
Chater-Diehl (Chater-Diehl et coll.,
2017
).
Les approches par gènes candidats, utilisant essentiellement des
modèles d’EPA chez la souris, sont basées sur la technique
d’immuno-précipitation de chromatine (ChIP) à l’aide d’anticorps
dirigés contre des marques d’histones suivi d’une amplification de
la région d’ADN du gène d’intérêt par qPCR (quantitative
Polymerase Chain Reaction ; on parle de ChIP-qPCR).
Globalement, les études de l’effet de l’EPA dans des périodes
prénatales chez la souris qui miment le premier et le second
trimestre chez la femme, soulignent le fait que des modifications de
l’expression ou de l’activité des acteurs impliquées dans le dépôt
de marques génétiques (DNMT [DNA-5mC-méthyl-transférase], HDAC...)
ne sont pas nécessairement corrélées avec une diminution ou une
augmentation parallèle de la marque épigénétique correspondante. En
d’autres termes, si ces changements contribuent peut-être à
l’altération des marques épigénétiques observées après EPA, ils n’en
sont pas la seule cause. Au contraire, on peut plutôt penser que
c’est la redistribution de ces acteurs à l’échelle du génome, sur
telle ou telle région, et ce de façon spécifique à chaque acteur,
qui va déterminer les remaniements des marques épigénétiques sur une
région donnée, selon des mécanismes qui restent à déterminer (voir
plus loin). De plus, pour chaque marque étudiée, le profil temporel
de ses altérations est différent. Enfin, ces changements de marques
épigénétiques ne sont pas nécessairement corrélés à des
modifications de l’expression des gènes qui les portent. On ne
dispose donc pas à l’heure actuelle d’une vue globale des
modifications d’histones qui procurerait une clé de lecture
interprétative de ces changements, en articulation avec ceux de
l’expression génique, et donc du phénotype résultant. Et cela, en
dépit du fait que des régions différentiellement méthylées (DMR)
sont associées à des gènes clés de neuro-développement : par
exemple, des gènes gouvernant le caractère « cellule souche » ou des
gènes homéotiques, ou des régions hétérochromatiniennes dont la
conformation fermée et dont le verrouillage de leur expression est
important pour la stabilité du génome. Il faudrait d’autre part que
le domaine s’astreigne à répéter des études utilisant les mêmes
protocoles dans divers laboratoires pour attester de la
reproductibilité des résultats.
À titre d’illustration concrète, des approches successivement
globales, puis de gènes candidats, dans la période postnatale qui
correspond, chez la souris, au troisième trimestre chez la femme et
est une phase active de synaptogenèse et de croissance cérébrale
(Chater-Diehl et coll., 2017
), on retiendra une étude fouillée qui
s’est intéressée chez la souris à
l’histone/lysine-méthyl-transférase KTM G9a et ses effets potentiels
sur l’apoptose après EPA. G9a, avec un autre membre de la famille,
GLP, est responsable de la di-méthylation de l’histone H3 sur les
résidus lysine K9 et K27 (H3K9me2 et HEK27me2). En accord avec
l’augmentation de l’expression de G9a en réponse à l’EPA dans
l’hippocampe et le cortex, les marques répressives H3K9me2 et
HEK27me2 sont augmentées par rapport à la quantité totale de H3
(qui, elle, diminue) de façon corrélée avec la mort neuronale
(Subbanna et coll., 2013
). Un prétraitement par un inhibiteur
spécifique de G9a réduit à la fois l’élévation des niveaux de
H3K9me2 et HEK27me2, la mort neuronale et les déficits de mémoire et
de reconnaissance sociale chez les souris adultes, issues de ce
protocole d’EPA (Subbanna et coll.,
2013
;
Subbanna et Basavarajappa, 2014
). Ils ont également confirmé sur un gène
candidat qui contrôle négativement le relargage de
neurotransmetteurs (CB1R, récepteur de type 1 des
endocannabinoïdes), une diminution de la marque H3K9me2 et d’une
augmentation de la marque H4K8ac sous l’effet de cet inhibiteur dans
l’hippocampe et le cortex (Chater-Diehl et coll.,
2017
;
Subbanna et coll., 2014
). Enfin, un antagoniste de CB1R et
l’inhibiteur de G9a annulent les effets de l’EPA sur les déficits en
plasticité neuronale, d’apprentissage et de mémoire, en lien avec
une restauration du profil épigénétique et de l’expression d’un gène
précoce de réponse de l’activité neuronale (
immediate-early
gene, Arc), profil qui est modifié par l’EPA (augmentation
de H3K9me2) (Subbanna et coll.,
2018a
;
Subbanna et coll., 2018b
). Cette étude suggère l’implication du
contrôle du niveau d’expression d’acteurs épigénétiques gérant les
modifications d’histones et ouvre la possibilité de leur
manipulation pharmacologique dans un but d’amélioration à la fois
des profils épigénétiques et des performances comportementales et
cognitives des souris ayant subi une EPA. D’autres travaux, comme
par exemple sur des gènes candidats impliqués dans la plasticité
synaptique ont montré une corrélation entre une de marques
d’histones activatrice (H3K14 acétylée, en ChIP-qPCR), une
hyperméthylation de l’ADN et une expression génique réduite (Dong et
coll., 2018
).
Cependant, lorsque de larges groupes de gènes sont explorés, les
variations des niveaux d’expression de l’enzyme ou de son activité
ne corrèlent ni avec la réduction, ni l’augmentation de la marque
épigénétique correspondante, et les altérations des marques
épigénétiques, bien que leur magnitude soit globalement dépendante
de la dose d’alcool, et ne s’accompagnent pas de modifications de
l’expression génique : ces études récentes se sont attachées à
l’effet à long terme de l’EPA chez la souris adulte sur l’ensemble
des promoteurs du génome (Chater-Diehl et coll.,
2016
) ou
à des modèles de cellules souches embryonnaires murines (Veazey et
coll., 2017
). Les modifications de marques d’histones induites par l’EPA ne
peuvent donc pas à elles seules être invoquées pour rendre compte
des traits liés au TCAF. Il est notable que, du moins à notre
connaissance, des études explorant directement l’accessibilité de la
chromatine et ses modifications sous l’effet de l’EPA, par ATAC-Seq,
par exemple (Buenrostro et coll.,
2013
;
Schang et coll., 2018
), n’aient pas encore été répertoriées dans
le domaine. En effet, les modifications des histones sont une chose,
leur impact sur la conformation chromatinienne en est une autre,
surtout si les « lecteurs » de ces marques sont perturbés dans leur
expression. En effet, leur régulation ou leur activité (par des
modifications post-traductionnelles, précisément) mettent en jeu les
mêmes acteurs que pour les histones. Il est possible aussi que les
marques déposées suite à l’EPA n’aient d’effets sur l’expression
génique que lors d’un stress ultérieur, ce qui demeure à tester.
Enfin, il se pourrait aussi que des modifications de la combinatoire
de facteurs de transcription sous l’effet de l’EPA, soient
responsables de modifications de l’expression génique qui peuvent
être observées à très grande distance temporelle de l’exposition –
combinatoire qui s’exercerait donc sur un paysage chromatinien
globalement robuste par rapport à l’EPA. De façon remarquable, il
existe une « mémoire » de l’EPA très précise, puisqu’elle se traduit
par des modifications de l’expression génique qui affectent des
programmes de transcription spécifiquement actifs au moment de
l’exposition (Kleiber et coll.,
2013
) :
les programmes de prolifération cellulaire chez la souris adulte
sont affectés de manière proéminente lorsque l’EPA s’est produite au
cours de l’équivalent du premier trimestre de grossesse chez la
femme, période pendant laquelle les progéniteurs neuraux se divisent
activement. Des atteintes de programmes gouvernant la migration
cellulaire (et donc neuronale) sont observées chez l’adulte quand la
période d’exposition coïncide avec celle qui mime le second
trimestre et qui se caractérise la migration de jeunes neurones pour
former les couches du cortex, en particulier. Enfin, lorsque l’EPA
s’effectue en période postnatale chez la souris, ce qui correspond à
la formation d’axones et de dendrites et à la synaptogenèse, on
observe une dérégulation majeure de gènes impliqués dans la
formation des synapses, le remaniement des réseaux neuronaux et la
plasticité cellulaire chez l’adulte. Les mécanismes moléculaires
sous-tendant la persistance spécifique de programmes d’expression
génique chez l’adulte restent donc encore obscurs et la question
demeure de la pertinence des modifications des histones en tant que
biomarqueurs potentiels, car il faudrait pourvoir explorer la
causalité entre ces marques et les phénotypes associés au TCAF.
Implication des microARN dans les perturbations
d’expression génique induites par l’EPA
Un autre niveau d’explication des discordances observées entre le
dépôt de marques d’histones anormales et les altérations de
l’expression génique après EPA est que les voies de régulations
majeures épigénétiques mises en œuvre au moment de l’EPA peuvent
opérer à un niveau post-transcriptionnel, en particulier à travers
la synthèse de miANR. Mir-10 est un candidat identifié par plusieurs
groupes (Miranda, 2012
; Laufer et coll.,
2013
),
qui régule des gènes homéotiques impliqués dans la migration
neuronale. Par ailleurs de nombreux miARN dont l’expression est
altérée par l’alcool régulent des gènes impliqués dans le
neurodéveloppement, soit à des stades précis (comme mir-10), soit à
tous les stades (comme mir-9). Leur fonctionnalité, en ce qui
concerne les effets de l’EPA, est soulignée par leur capacité à
affecter de nombreux gènes dont des perturbations de l’expression
sont répertoriées dans des modèles de maladies
neurodéveloppementales ou neuropsychiatriques (par exemple la
microcéphalie, le déficit intellectuel). La délétion de plusieurs
mir-9 conduit en effet à un phénotype mimant le TCAF chez la souris.
Il est à noter que l’expression de nombreux miARN est elle-même sous
contrôle épigénétique, et donc sensible à l’EPA (Miranda,
2012
).
Identification de signatures épigénétiques du méthylome
de l’ADN par approches non-biaisées à l’échelle du
génome
Nous nous concentrerons sur les études concernant majoritairement
l’analyse du méthylome de l’ADN à grande échelle (échelle du génome ou
d’une partie « capture » du génome). Aux facteurs de variabilité liés au
modèle d’étude, au mode d’administration de l’alcool et aux fenêtres de
temps, se rajoute le fait que les méthodes d’explorations du méthylome
de l’ADN sont également très diverses (figure 5.1
; Schang et coll.,
2018
). Enfin,
les méthodes bioinformatiques et biostatistiques utilisées pour
l’identification des DMR varient. Il est donc impossible d’établir de
manière rigoureuse une liste pertinente des gènes ou de régions
concernées dans les perturbations épigénétiques induites par l’EPA. Il y
a plus de 28 millions de CpG dans le génome humain et on estime que
70-80 % d’entre elles peuvent être méthylées, même si la grande majorité
ne l’est pas. 45 000 environ sont situées dans des îlots CpG
(figure 5.1
). Ces chiffres
illustrent la complexité rencontrée par les recherches. Des tendances
néanmoins se dégagent qui sont en faveur de la détection de biomarqueurs
d’EPA et de leur utilisation future en prévention ou suivi.
D’une manière générale, on n’observe ni une hyperméthylation globale du
génome, ni une hypométhylation gobale en réponse à l’EPA, mais une
redistribution de la méthylation à l’échelle du génome : certaines
régions sont hyperméthylées, tandis que d’autres sont hypométhylées et
ce, même à distance temporelle de l’EPA (chez l’adulte : Kleiber et
coll., 2013
;
Laufer et coll., 2013
; Lussier et coll.,
2018b
). Ces
résultats suggèrent que les DNMT sont redistribuées sur d’autres régions
que leurs sites physiologiques par des modes de recrutement qui restent
encore à éclaircir.
Lorsque l’on étudie le méthylome de l’ADN à l’échelle du génome (et non
seulement sur des « captures » d’une partie du génome), on observe que
de nombreuses DMR semblent situées dans des régions dites
« intergéniques », c’est-à-dire à distance des gènes et de leurs régions
régulatrices proximales (Lussier et coll.,
2018
). Ces régions
comportent potentiellement des éléments régulateurs de type
enhancers dont l’activité pourrait être perturbée par l’EPA.
Mais il est souvent très difficile à l’heure actuelle de savoir quel(s)
gène(s) précisément elles régulent (voir plus loin) et donc de tester
l’impact fonctionnel de ces modifications. Cependant, les nombreux sites
de facteurs de transcription qu’elles contiennent, ainsi que leur
identification, pourrait éclairer de façon majeure la manière dont les
DNMT sont recrutées à l’échelle du génome et dont on pourrait manipuler
leur redistribution dans un but de normalisation. En effet, dans les
régions proximales mieux connues, de nombreuses DMR correspondent aussi
à des régions régulatrices riches en sites de facteurs de transcription
(Laufer et coll., 2013
; Khalid et coll.,
2014
; Laufer
et coll., 2017
;
Lussier et coll., 2018a
) dont en sites de liaison du facteur CTCF qui
a également un rôle d’« insulateur », car il borne des régions
chromatiniennes de conformations différentes et est ainsi un élément clé
de la structure fonctionnelle de l’épigénome et de la régulation de
l’expression des gènes. La méthylation de tels sites de facteurs de
transcription peut affecter leur liaison à l’ADN et remodeler ainsi
l’expression génique.
Les DMR (en particulier celles caractérisées par une hyperméthylation)
sont associées à des gènes de neuro-développement (Kleiber et coll.,
2014
; Laufer
et coll., 2017
;
Lussier et coll., 2018a
). Elles ont été aussi souvent identifiées dans
les explorations du méthylome d’autres pathologies
neurodéveloppementales ou neuropsychiatriques, (ex : la famille de
protéines synaptiques SHANK, (Bourgeron,
2015
) :
troubles de l’anxiété, syndrome épileptique, troubles du spectre
autistique, trouble de développement profond (un ensemble de cinq
pathologies caractérisées par un retard de fonctions cérébrales
élémentaires, (dont la communication et la socialisation) et troubles
liés à des substances (Portales-Casamar et coll.,
2016
). Cela
est en faveur de l’identification de marques de méthylation de l’ADN
comme futurs biomarqueurs.
L’exploration des impacts épigénétiques ne concerne pas que l’exposition
in utero et les TCAF. En effet, des études suggèrent un effet
pré-conceptionnel de la consommation de l’alcool par le père (Beeler et
coll., 2019
;
Chang et coll., 2019
) – soit, potentiellement, par des marques de
méthylation de l’ADN anormales, soit par la perturbation du contenu en
ARN non-codants dans le sperme – ainsi qu’un impact péri-conceptionnel
par la mère (Lucia et coll., 2019
).
Signatures épigénétiques induites par l’EPA
comme
biomarqueurs potentiels
Modifications des histones comme biomarqueurs
potentiels
Idéalement, un biomarqueur doit appartenir à la chaîne causale d’un
processus biologique qui, endommagé, participe à l’étiologie de la
pathologie et n’est pas lié à des facteurs inconnus agissant lors de
l’exposition. Les modifications chimiques des histones impliquées
dans le TCAF, sont donc de faibles candidats, même en ce qui
concerne la marque K3K9me3 qui présente une robustesse dans le temps
après EPA, qui est identifiée dans diverses études, et qui est
techniquement accessible (Chater-Diehl et coll.,
2017
).
Par ailleurs, on distingue des biomarqueurs d’exposition, destinés à
établir des prédictions, et des biomarqueurs de pathologies,
utilisés pour faciliter le diagnostic. La relative corrélation entre
les perturbations de marques et la sévérité du FASD établie dans des
modèles animaux (Veazey et coll.,
2015
)
pourrait faire de certaines marques d’histones opérant sur des gènes
spécifiques, des biomarqueurs de l’exposition et de son ampleur,
mais elle ne serait en aucun cas prédictive des effets
comportementaux qui, d’un patient à l’autre, peuvent varier
considérablement. Les développements spectaculaires de l’analyse de
l’épigénome par imagerie, en termes de conformation de la chromatine
(Chen et coll., 2016
) ou du décodage de la combinatoire de
modifications portées par un nucléosome, à l’échelle de la molécule
unique (Shema et coll., 2016
) pourraient à l’avenir permettre une
exploration plus directe et plus pertinente de l’accessibilité de la
chromatine et des marques d’histones.
Signatures de méthylation de l’ADN comme
biomarqueurs potentiels
La méthylation de l’ADN est une marque techniquement plus facile
d’accès puisqu’il est plus facile d’isoler et de conserver l’ADN
méthylé que d’explorer les marques d’histones par ChIP-qPCR ou
ChIP-Seq (ChIP, suivie de séquençage du génome à haut débit). Les
aspects fonctionnels des marques de méthylation de l’ADN sont
également mieux appréhendés car mieux corrélés à l’expression
génique, comme nous l’avons vu.
Les méthodes actuelles d’exploration du méthylome de l’ADN n’ont
considéré, pour la plupart, qu’une partie du génome (« captures »)
et, en particulier, la couverture des éléments
enhancers de
régulation des gènes est actuellement faible dans ces technologies.
Or, des DMR ont été identifiées dans des régions inter-géniques très
riches en
enhancers (Lussier et coll.,
2018
), mais aussi
les corps de gènes ou des régions transcrites, mais non traduites
(5’UTR), et l’on peut penser qu’une exploration plus représentative
des différentes régions du génome devrait conduire à
l’identification de nouvelles DMR d’intérêt pour la détermination de
biomarqueurs. Une très récente étude à l’échelle du génome s’appuie
sur une cohorte très bien caractérisée pour des sous-phénotypes du
TCAF (Cobben et coll., 2019
). Un enrichissement des DMR dans les
régions correspondant au corps des gènes et au démarrage de la
transcription a été observé (Lussier et coll.,
2018b
).
Quatre DMR robustes ont été identifiées, associées au moins à un
sous-phénotype, dont l’une (
NECAB3) est partagée avec les
travaux de Portales-Casamar et coll.
(2016
) et
de Lussier et coll. (2018a
) et présente donc un potentiel de
biomarqueur. Le faible chevauchement de ces trois études repose à la
fois sur les méthodes d’approches différentes pour explorer le DNA
méthylome, mais aussi sur la manière dont les cohortes ont été
construites (stratification des patients par sous-phénotypes, en
particulier).
Les études de Laufer et coll. (2013
), Chater-Diehl et coll.
(2016
),
Portales-Casamar et coll. (2016
) et Lussier et coll.
(2018b
)
ont mis en évidence, dans des cohortes d’enfants TCAF, des DMR
associées à des gènes importants pour diverses étapes et pathologies
neurodéveloppementales, dont, de façon remarquable :
i) le
large groupe (
clusters) de gènes codant les protocadhérines
dont les rôles clés dans le développement neural se sont récemment
renforcés et étendus (Toyoda et coll.,
2014
;
Aran et coll., 2016
; El Hajj et coll.,
2016
;
Molumby et coll., 2017
) ;
ii) des gènes soumis à
l’empreinte parentale, c’est-à-dire qui montrent une expression
spécifique à partir d’un seul des deux allèles, selon son origine
parentale, sur la base de la méthylation, soit de l’allèle maternel,
soit de l’allèle paternel – l’autre étant maintenu épigénétiquement
silencieux. De façon intéressante, 30 % des gènes soumis à
l’empreinte expriment des ANR non-codants, dont la dérégulation est
impliquée dans les désordres neuro-développementaux, tels que ceux
issus du locus
SNRPN-UBE3A exprimé spécifiquement dans les
neurones et associé au Syndrome de Prader-Willi (LaSalle et coll.,
2015
).
Cette expression, étroitement régulée par des mécanismes
épigénétiques, est cruciale pour le neuro-développement (Davies et
coll., 2008
).
Les deux «
hots-spots » constitués par les groupes de gènes
de protocadhérines, d’une part, et les gènes soumis à l’empreinte,
d’autre part, ont été aussi identifiés dans des analyses du
méthylome de l’ADN dans des modèles murins de FASD à distance de
l’exposition (chez l’adulte) et liés à des modifications de
l’expression des ARN (Laufer et coll.,
2013
;
Laufer et coll., 2015
). Ils présentent donc un potentiel de
biomarqueurs important.
De façon intéressante, et qui pourrait permettre de discriminer
potentiellement à l’avenir le type d’exposition dû à la consommation
d’alcool par les parents, les études d’exposition
pré-conceptionnelle d’origine paternelle ne montrent pas de
modifications de la méthylation de l’ADN ou de l’altération de
l’empreinte dans le sperme, mais une expression différentielle de
gènes soumis à l’empreinte dans le placenta des fœtus qui en sont
issus (Chang et coll., 2017
).
L’hétérogénéité des cohortes TCAF recrutées jusqu’à présent pour des
études du méthylome de l’ADN (données démographiques de sexe, âge,
ethnicité...) (Laufer et coll.,
2015
;
Chater-Diehl et coll., 2016
; Portales-Casamar et coll.,
2016
;
Lussier et coll., 2018a
) et leur taille très restreinte
constituent un frein à l’identification de marqueurs robustes
pouvant s’appliquer à des populations étendues. Le succès de deux
études combinant des méthodes de détection d’événements de
méthylation de l’ADN à des approches d’intelligence artificielle
(
machine learning) pour la détection de l’exposition
prénatale au tabac est néanmoins prometteur à ce titre pour l’EPA
(Joubert et coll., 2012
; Ladd-Acosta et coll.,
2016
).
Perturbations de l’épigénome, potentiel en
biomarqueurs et thérapies
et troubles de l’usage de l’alcool chez
l’adulte
Nous ne décrirons pas en détail les explorations de l’épigénome liées
aux consommations à risque d’alcool chez l’adulte (consommation,
sevrage) et leur lien avec l’addiction ou d’autres pathologies
neuropsychiatriques. Ces investigations en sont encore à leur début,
et des discordances apparaissent entre diverses études, dues à la
taille modeste et à la définition des cohortes. Des modifications de
la méthylation de l’ADN ou d’hydroxy-méthylation (Koller et coll.,
2019
) ont
été trouvées dans des tissus périphériques, dans les régions
promotrices de gènes, reliées aux consommations à risque, que ce
soit durant la période de consommation ou de sevrage, et en lien
avec des phénomènes de neuro-adaptation. Ces événements de
méthylation pourraient ainsi sous-tendre un rôle complexe dans le
risque de troubles liés à l’usage de l’alcool persistants (Zhang et
Gelernter, 2017
) et sont localisées, par exemple, dans
des séquences répétées du génome, de régions régulatrices de gènes
du métabolisme de l’alcool, du stress oxydatif et de la réponse
immunitaire (aldéhyde-déshydrogénases, cytochrome P450,
TLR4
Toll-like Receptor 4 [Ureña-Peralta et coll., 2018
]...), et
dans les gènes du transporteur de la dopamine (
DAT), du
récepteur de la dopamine (
DRD2 ; Hagerty et coll.,
2020
;
Hill et Sharma, 2019
), du sous-type de Récepteur
N-méthyl-D-aspartate 2b (
GRIN2B), du facteur de croissance
NGF (
nerve growth factor), du récepteur µ opioïde
(
OPRM1), du transporteur de la sérotonine
(
5HTT/
SLC6A4), avec pour ce gène une cohérence
avec des modifications de l’expression. Des interactions génome x
épigénome ont aussi été mises en évidence : des polymorphismes sur
des nucléotides CpG (SNP-CpG) qui affectent donc les possibilités de
méthylation (Zhang et Gelernter,
2017
).
Le développement des explorations à l’échelle du génome a confirmé
l’importance de perturbations de la méthylation de l’ADN sur le
promoteur du gène
GDAP1 (
Ganglioside induced
Differentiation Associated Protein 1) par ailleurs associé à
la maladie de Charcot-Marie-Tooth (Zhang et Gelernter,
2017
) et
révélé aussi des sites CpG exploitables par PCR comme futurs
biomarqueurs d’exposition (Philibert et coll., 2018
). Des variations
d’expression de miARN ont également été identifiées qui en lien avec
des marqueurs génétiques (SNP), pourraient prédire la prédisposition
aux consommations à risque d’alcool (Rudra et coll.,
2018
).
Les enjeux pour la détection de biomarqueurs de ces troubles,
d’identification de marques « miroirs » dans les tissus
périphériques (Berg et coll., 2018
; Clark et coll.,
2018
;
Perrier et coll., 2019
) sont les mêmes que pour le TCAF. Ils sont
moins complexes que pour le TCAF, néanmoins, puisque qu’ils
s’adressent à l’adulte, et concernent en particulier : 1) la
détection des consommations à risque d’alcool ; 2) les problèmes
liés à la prescription de médicaments « épigénétiques » par rapport
aux problématiques
in utero ou en période périnatale, du
fœtus, de l’enfant ou de l’adolescence (voir plus loin).
Perspectives et freins pour l’identification et l’usage
de futurs biomarqueurs de TCAF
Interprétation fonctionnelle du dépôt de marques
épigénétiques aberrantes après EPA
Les études menées chez l’adulte qui ont été exposés à l’alcool durant
la vie fœtale soulèvent la question suivante : les marques
épigénétiques aberrantes observées sont-elles directement dues à
l’EPA ou correspondent-elles plutôt au dépôt de marques
épigénétiques en réponse à des défauts de fonctionnement des
circuits neuronaux engendrés
a posteriori par cette
exposition ? Peu d’études ont été entreprises chez le nouveau-né
exposé, pour des raisons évidentes de difficultés d’accès au
matériel biologique que constitue le cerveau. Des changements
épigénétiques globaux ont été néanmoins décelés
post mortem
dans le noyau des cellules neurales (« nerveuses », neurones,
astrocytes, oligodendrocytes) dans le cerveau de fœtus et d’enfants,
et bien qu’ils soient d’interprétation délicate, suggèrent que des
études approfondies de l’épigénome devraient être entreprises pour
cartographier ces marques épigénétiques qui semblent se déposer à
très courte distance temporelle de l’exposition (Jarmasz et coll.,
2019
).
De plus, la fonctionnalité de ces perturbations reste obscure dans de
nombreux cas. D’une part, un grand nombre d’études présentent des
résultats (DMR) sur cerveau entier, ce qui pose le problème de leur
interprétation par rapport aux phénotypes pertinents pour le TCAF
touchant à la prolifération, la différenciation neuronale,
l’activité et la plasticité synaptique ou au comportement. La
validation moléculaire de l’impact de ces modifications sur
l’activité de gènes pertinents reste à démontrer dans l’immense
majorité des cas. Les approches appropriées devraient cibler la
population cellulaire d’intérêt, et la ou les régions du génome
modifiées, dans un but rectifier la signature épigénétique par des
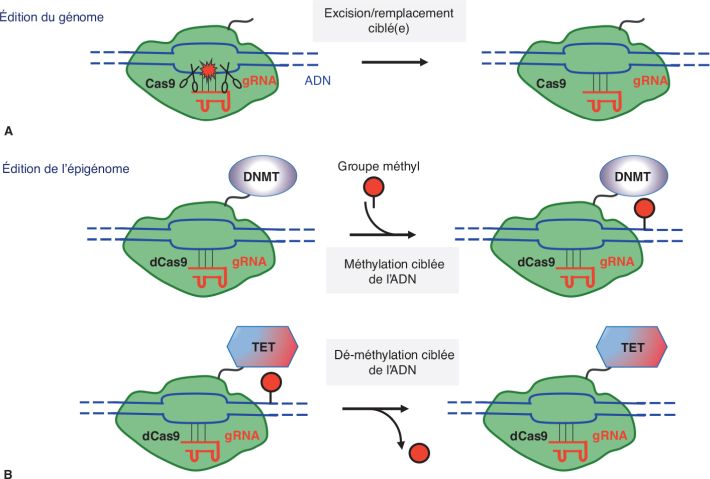
techniques dérivées de l’édition des génomes
(figure 5.2
; Holtzman et
Gersbach, 2018
), afin d’en explorer l’impact positif sur l’amélioration ou la
restauration d’un niveau physiologique de l’expression génétique et,
le cas échéant, du phénotype cellulaire ou de la fonction neuronale.
Cette technologie laisse intacte la séquence de l’ADN, mais cible,
sur une région précise du génome, un acteur épigénétique capable de
restaurer les marques épigénétiques (par exemple une HAT/KAT ou une
HDAC/KDAC). On pourrait ainsi explorer l’impact de la restauration
de cette/ces marque(s) sur l’expression génique et les phénotypes
liés au TCAF. Cette démonstration de la signification biologique
d’une DMR permettrait donc de la retenir en tant que biomarqueur
potentiel.
De nombreuses DMR sont situées dans des régions inter-géniques –
c’est-à-dire à distance des gènes. Comme nous l’avons vu, de
nombreuses régions régulatrices de l’activité des gènes
(
enhancers) sont localisées dans ces régions
inter-géniques, et il est bien difficile à l’heure actuelle
d’attribuer les événements de méthylation, identifiés dans une DMR,
aux perturbations de l’expression d’un gène donné, celui-ci pouvant
se trouver à grande distance de la DMR, voire même sur un autre
chromosome. Les nouvelles approches de cartographie de
l’architecture tridimensionnelle du génome (technologie de Hi-C,
dérivée de la Chromosome Conformation Capture 3C) devraient
contribuer à attribuer une DMR à un ou plusieurs gènes, à travers
l’identification de domaines chromatiniens appelés
«
Topologically Associated Domains » (TADs) et de régions
plus restreintes (sous-TADs) et ce, également, dans des approches en
cellule unique et non seulement en populations cellulaires (Lee et
coll., 2019
).
Le fait que des inhibiteurs d’acteurs épigénétiques aient des effets
bénéfiques de réduction des défauts neuro-développementaux et des
endophénotypes liés aux TCAF pourrait aussi passer par la
perturbation de modifications post-traductionnelles de protéines
non-histones, comme des facteurs de transcription ou d’autres types
de régulateurs dont l’activité est aussi contrôlée par
l’acétylation, la méthylation ou d’autres modifications chimiques.
Comme nous l’avons déjà évoqué, un remaniement de la combinaison de
tels facteurs de transcription dans des régions chromatiniennes dont
l’accessibilité serait inchangée pourrait aussi rendre compte des
perturbations d’expression génique observée en réponse à l’EPA,
ainsi que de la modification de l’expression de miARN.
Interaction entre gène et
environnement/épigénétique
La sensibilité du fœtus à l’alcool repose non seulement sur les modes
d’EPA (doses, chronicité, consommation massive d’alcool sur une
courte durée...) mais aussi sur sa propre architecture génétique et
de celle de ses parents : présence de variants ou de combinaisons de
variants génétiques, favorisant une vulnérabilité aux TUA, chez la
mère ou le père, et aux défauts neuro-développementaux susceptibles
d’être engendrés par l’EPA, chez le fœtus. Ce concept « gène x
environnement » est particulièrement pertinent dans le cas de
maladies neuropsychiatriques dont la composante génétique est faible
(comme la dépression majeure, ou la vulnérabilité aux addictions).
Les développements spectaculaires du séquençage à haut-débit des
génomes et la création de très grandes cohortes ont récemment permis
d’identifier des variants associés à un risque plus élevé de
développer une dépression majeure (CONVERGE consortium,
2015
) ou
des consommations à risque d’alcool (Liu et coll.,
2019
).
Cela pourrait à terme avoir des répercussions positives sur la
capacité à cerner la vulnérabilité au TCAF, si appliqué aux parents
ou même au fœtus. À l’avenir, des études sur le TCAF pourraient
potentiellement révéler l’existence de variants dans des gènes
codant des acteurs épigénétiques, comme c’est le cas, pour la
dépression majeure, de la HDAC SIRT1, un intégrateur important du
métabolisme (CONVERGE consortium,
2015
).
Plus encore, une étude sur la prédiction des causes de mortalité
(toutes confondues) suggère que les informations épigénétiques
seraient potentiellement plus informatives que les génétiques (Zhang
et coll., 2017
), incitant fortement à poursuivre les études des remaniements de
l’épigénome en réponse à l’EPA dans la mesure où ils pourraient
permettre d’identifier des individus à risque et de permettre la
mise en place de stratégies de prévention ou de suivi.
Accès aux signatures épigénétiques : le parallèle
entre cerveau et tissus périphériques
L’identification de biomarqueurs épigénétiques pertinents
caractéristiques du TCAF est freinée par la grande difficulté, sinon
l’impossibilité d’effectuer des biopsies du cerveau. Pour contourner
cette limitation, les prélèvements de tissus périphériques (cellules
sanguines nucléées, épithélium buccal [récolté par frottis] ou
olfactif...) peuvent constituer une alternative, à condition qu’ils
représentent un bon miroir des altérations de l’épigénome de la
région cérébrale d’intérêt. Les cellules buccales présentent
l’intérêt d’avoir la même origine embryonnaire ectodermique que les
cellules du cerveau. Des DMR spécifiques de l’EPA ont été
identifiées à la fois dans l’hypothalamus et les leucocytes de rat
et montrent des changements dans la même direction dans ces deux
types de tissus (Lussier et coll.,
2018b
).
Ces DMR contiennent de nombreux sites potentiels de facteurs de
transcription et touchent des gènes impliqués dans la fonction
immunitaire, le remodelage épigénétique, le métabolisme et la
signalisation hormonale. L’une d’elles concerne le gène codant le
récepteur à la dopamine D4,
DRD4, qui a été trouvée également
dans les échantillons d’épithélium buccal de cohortes TCAF
(Fransquet et coll., 2016
; Portales-Casamar et coll.,
2016
).
Des événements de méthylation de l’ADN placentaires ont montré une
association avec l’EPA. Ils ont été identifiés sur des régions
répétées du génome dans des régions proches de site de démarrage de
la transcription, et riches en site de facteurs de transcription
pour des processus importants (modulation immunitaire et
métabolisme) qui ouvre également une piste (Loke et coll.,
2018
).
Des miARN maternels circulants, présents à des taux élevés en milieu
et fin de grossesse, ont été identifiés dans des études visant à
prédire le devenir des enfants après EPA (Balaraman et coll.,
2016
;
Tseng et coll., 2019a
; Tseng et coll.,
2019b
) et
représentent également des biomarqueurs potentiellement
intéressants.
Stratégies d’avenir d’identification de
biomarqueurs
La combinaison d’approches de séquençage à très haut-débit (à
l’échelle de la cellule unique ou non), des techniques dérivées de
l’édition du génome et de l’épigénome (figure 5.2
) et de l’intelligence artificielle, est
susceptible de faire apparaître de nouvelles voies de dérégulation
proéminentes dans le TCAF.
Par exemple, une étude transcriptomique utilisant des approches
algorithmiques d’apprentissage automatique non supervisé
(
unsupervised machine learning) dans un modèle d’embryon
de poulet a mis en évidence des miARN co-régulant plusieurs voies de
régulation. Certaines étaient connues, d’autres ont été révélées
grâce à une réduction de la complexité des données (Al-Shaer et
coll., 2019
).
Cette combinaison d’approches de pointe peut s’appliquer à des
modèles cellulaires ou organoïdes (Luo et coll.,
2016
)
issus de patients TCAF (cellules pluripotentes induites
différenciées en neurones par exemple) et être couplée à de
l’imagerie à haut contenu-haut débit qui, faisant également appel à
l’intelligence artificielle, constitue une stratégie de choix pour
la découverte de nouvelles molécules à potentiel thérapeutique par
des cribles à (très) haut débit.
Signatures épigénétiques et diagnostic précoce de
FASD
Un biomarqueur n’a pas de valeur pris isolément, mais uniquement en
combinaison avec d’autres biomarqueurs et à la lumière d’un tableau
clinique global. Associé à celui-ci, et à des marqueurs biochimiques
métaboliques (Lecuyer et coll., 2017
; Andreu-Fernández et coll.,
2019
),
d’imagerie cérébrale poussée (Chabenne et coll.,
2014
;
Paolozza et coll., 2017
), de phénotypage comportemental
(l’enregistrement des mouvements de l’œil : Paolozza et coll.,
2014a
;
Paolozza et coll., 2014b
; Paolozza et coll.,
2015
; Zhang
et coll., 2019
),
des tests psychomoteurs et neuropsychiatriques, l’identification de
marques épigénétiques pourrait conduire à un diagnostic plus précoce de
l’EPA et à la prise en charge de jeunes patients pour une remédiation
non-médicamenteuse ou médicamenteuse. De façon notable, le dimorphisme
sexuel observé pour certains biomarqueurs moléculaires ou
comportementaux (Lee et Rivier, 1996
; Weinberg et coll.,
2008
;
Paolozza et coll., 2015
; Loke et coll.,
2018
),
pourrait également concerner de futurs marqueurs épigénétiques, avec le
biais actuel suivant : la majorité des études épigénétiques sur l’animal
ont concerné des mâles pour s’affranchir des variations potentiellement
liées au cycle œstral chez les femelles (Lussier et coll.,
2017
). Plus
le diagnostic et la prise en charge seront précoces, meilleures seront
les chances d’améliorer les capacités cognitives et comportementales du
jeune patient et sa trajectoire de vie.
Épigénétique et perspectives
thérapeutiques
La combinaison d’approches de pointe (génome x épigénome, technologies
d’édition de l’(épi)génome [figure 5.2
], intelligence artificielle, cellules pluripotentes issues de
patients TCAF ou ayant subi une EPA et différenciées en cellules
neurales ou en organoïdes cérébraux) peut être non seulement appliquée à
l’identification de biomarqueurs, mais également être couplée à de
l’imagerie à haut contenu-haut débit qui, faisant également appel à
l’intelligence artificielle, constitue une stratégie de choix pour la
découverte de nouvelles molécules thérapeutiques.
Perspectives d’intervention
médicamenteuse
Les marques aberrantes observées après EPA touchent donc des gènes
impliqués dans la performance des fonctions neuronales et un certain
nombre d’études suggère qu’elles sont associées à un
dysfonctionnement cérébral – même s’il reste à le démontrer au
niveau moléculaire, en utilisant des outils d’édition de l’épigénome
(figure 5.2
). Des compléments
alimentaires à base de choline semblent avoir des effets bénéfiques,
mais uniquement chez de très jeunes enfants (2-5 ans), suggérant un
effet sur le développement cérébral (Wozniak et coll.,
2015
) et
indiquant que ces marques épigénétiques sont potentiellement
réversibles. Il est tentant d’imaginer qu’en jouant sur l’activité
des enzymes impliquées dans leur dépôt sur le génome, on pourrait
effacer ces marques et retourner à une conformation normale de la
chromatine, et de l’expression de ces gènes. Le problème n’est pas
simple car il ne suffit pas d’exciser les marques aberrantes, mais
aussi de favoriser le dépôt des marques normales, physiologiques. La
balance et le dialogue existant entre les machineries de méthylation
de l’ADN et de dépôt de marques répressives/activatrices sur les
queues des histones est un levier intéressant. Des « médicaments
épigénétiques » (
epidrugs) sont d’ailleurs déjà utilisés en
clinique dans le traitement de certains cancers, et depuis fort
longtemps en psychiatrie (tel que l’acide valproïque ; Sharma et
coll., 2005
) : c’est le cas des inhibiteurs de HDACs qui, en empêchant la
désacétylation d’histones, favorisent le retour à une conformation
ouverte de la chromatine et à une expression physiologique des gènes
(figure 5.1
). Des exemples
encourageants sont procurés par des études effectués dans les
modèles animaux adultes, dans un contexte d’addiction (Drissi et
coll., 2020
).
Cependant, la multiplicité des HDAC, combinée aux difficultés
d’identifier des inhibiteurs spécifiques des différents membres des
HDAC, freinent le développement thérapeutique. De plus, ces
inhibiteurs agissent globalement sur le génome et sont susceptibles
de déréguler d’autres gènes qui n’ont pas été perturbés par l’EPA,
avec un risque de bouleversement majeur de la fonction neuronale.
Enfin, le mode d’administration systémique de ces molécules implique
qu’elles affectent d’autres tissus et cellules que ceux et celles
que l’on veut cibler (ici le cerveau, dans des régions précises et
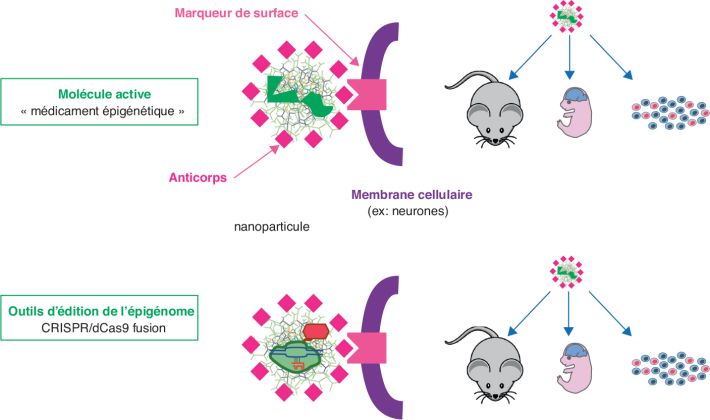
des populations cellulaires précises). L’utilisation de
nanoparticules capables de passer la barrière hématoencéphalique et
de cibler spécifiquement une population cellulaire donnée pourrait à
l’avenir contribuer à résoudre ce problème, car on peut les associer
à des anticorps spécifiques de marqueurs cellulaires
(figure 5.3
).
Les techniques dérivées de l’édition de l’épigénome, couplées à
l’utilisation de ces nanoparticules, sont susceptibles de diriger
une molécule thérapeutique sur une cible génique (ou une combinaison
de cibles géniques) (figures 5.2
et 5.3
). Encapsulées dans des
nanoparticules
ad hoc, ces acteurs couplés à ces outils
d’édition, pourraient modifier de façon « chirurgicale »
l’accessibilité de la chromatine dans des gènes clés et rectifier
l’expression de gènes d’importance pour le neuro-développement ou
les fonctions neuronales, dans une sous-population cellulaire
ciblée. De telles approches ont déjà été tentées pour des maladies
neurodéveloppementales (Liu et coll.,
2016b
;
Liu et coll., 2018
; Wang et Jiang,
2019
).
La caractérisation des modifications de l’épigénome dans le TCAF et
l’identification de gènes à cibler de façon privilégiée en fonction
de leur rôle dans le neurodéveloppement rend possible d’envisager ce
genre d’approche pour ce spectre de pathologies. Ces nouvelles
technologies laissent donc entrevoir des solutions thérapeutiques
« à façon », dans le futur, mais la route est encore longue entre
les preuves de concept chez l’animal et leur mise en place chez
l’homme et encore plus chez l’enfant.
Outre ces aspects techniques, comme nous l’avons vu, le développement
du cerveau est particulièrement contrôlé par des mécanismes
épigénétiques ; la diversification et la dynamique très particulière
des marques de méthylation de l’ADN de la naissance à l’adolescence,
touche plus particulièrement les neurones, mais aussi les cellules
gliales, et engendre une complexité inédite de leur combinaison et
de leur interprétation, par les machineries épigénétiques, sur
l’expression des gènes. Ces caractéristiques rendent très complexe
et hasardeuse la perspective de la manipulation du méthylome de
l’ADN, en particulier, dans le cerveau de nouveau-nés, de jeunes
enfants et d’adolescents.
De plus, dans une population cellulaire neurale donnée, toutes les
cellules ne vont pas être touchées de la même manière par l’EPA. La
réponse à l’EPA présente un aspect fortement stochastique. Le
cerveau en développement présente en effet une variabilité
physiologique entre cellules (Pollen et coll.,
2014
),
même dans une population cellulaire bien caractérisée que l’on peut
identifier à l’aide de marqueurs (ex. : progéniteurs neuraux ;
jeunes neurones de couches spécifiques du cortex ; cellules gliales
et non neurales ; microglie d’origine immunitaire). Cette
variabilité procure vraisemblablement, au cerveau en développement
et adulte, une adaptabilité aux exigences du fonctionnement et de la
plasticité neuronale, ainsi qu’en réponse à des variations de
l’environnement. Cependant, cette variabilité entre cellules est
exacerbée par l’EPA et c’est dans les cellules les plus
« déviantes » que l’expression des gènes qui contrôlent le
neuro-développement est la plus dérégulée (Hashimoto-Torii et coll.,
2014
;
Ishii et coll., 2017
). Idéalement, il faudrait pouvoir cibler
uniquement cette sous-population cellulaire vulnérable sans
perturber celles qui ont adopté un destin cellulaire normal. On
conçoit donc que, même si les outils moléculaires existent pour
faire des « frappes » chirurgicales en vue de restaurer les marques
épigénétiques anormales, le chemin soit encore long.
Remédiation
non-médicamenteuse
Des travaux ont répertorié les interventions non-médicamenteuses qui
visent à améliorer l’état des jeunes patients TCAF. Ces
interventions incluent : des stratégies d’éducation et
d’apprentissage (utilisant ou non les jeux vidéo et la réalité
virtuelle), la thérapie du contrôle cognitif (visant à rétablir une
hiérarchie satisfaisante entre les informations provenant de
l’environnement extérieur et intérieur, et la connexion entre la
pensée et l’action par auto-observation et autorégulation), la
thérapie langagière et linguistique, les interventions précoces, en
mathématiques, et l’entraînement par répétition pour la mémoire, et
les stratégies sociales et comportementales. Quelques études bien
randomisées avec des cohortes contrôles qui s’intéressent à des
déficits fonctionnels spécifiques semblent indiquer que ces
stratégies peuvent apporter effectivement des améliorations (Peadon
et coll., 2009
). Des interventions, visant à former les parents (mères
consommant des substances psychotropes) ou les soignants, réduisent
le risque de dépression chez l’enfant, la consommation de drogues ou
d’alcool chez les mères, le cas échéant, et le comportement de
l’enfant. Des thérapies parent-enfant montrent également des effets
bénéfiques sur le comportement de l’enfant et le stress des parents.
Certaines sont destinées aux adultes TCAF ou aux adolescents TCAF et
leurs parents et réduisent la consommation de drogues et d’alcool,
la prise en charge des responsabilités familiales, le recours aux
services médicaux et de santé mentale (Petrenko et Alto,
2017
).
Même si là encore, tout reste à démontrer en ce qui concerne le TCAF,
on peut supposer que ces interventions, à travers la stimulation
d’une activité neuronale améliorée agisse en partie sur des marques
épigénétiques, comme cela est suggéré par des études chez l’adulte
concernant les effets bénéfiques de l’exercice physique (Chen et
coll., 2018
).
Conclusion
L’étude de l’épigénome, de sa robustesse ou de sa plasticité, de la
question du dépôt précoce de marques épigénétiques suite à l’exposition
et de leur persistance à distance temporelle, ainsi que de l’impact
fonctionnel de ces perturbations, est donc une piste à poursuivre de
façon privilégiée, que ce soit suite à une EPA, sur le cerveau de
l’enfant et de l’adulte, ou suite à une consommation chez l’adulte. Il
est tout aussi important de favoriser en parallèle la constitution de
larges cohortes bien phénotypées (données démographiques de sexe, d’âge,
d’ethnicité, de mode de recrutement d’alcoolisation ou de sevrage...).
La réversibilité du dépôt de marques épigénétiques procure un espoir
thérapeutique certain. Elle s’articule avec une perspective de couplage
avec des outils issus de l’édition du génome et d’administration de
molécules à potentiel thérapeutique ciblées sur des sous-populations
cellulaires via l’utilisation de nanoparticules (et non
systémique). La caractérisation de ces sous-populations cellulaires,
grâce aux approches sur cellule unique en plein essor (transcriptomiques
et épigénomiques), est une voie prometteuse. Le développement de
l’intelligence artificielle, que ce soit pour l’analyse de
transcriptomes ou d’épigénomes, mais aussi en imagerie à haut contenu et
haut débit (par exemple l’imagerie de la conformation de la chromatine),
est susceptible d’ouvrir des brèches remarquables dans notre
compréhension des mécanismes sous-tendant le TCAF et la découverte de
nouvelles voies thérapeutiques. Cependant, dans l’état actuel de nos
connaissances (datant d’une ou deux décennies), le traitement
médicamenteux d’enfants TCAF soulève un grand nombre de questions et la
prévention de la consommation d’alcool pendant la grossesse et en
période de préconception reste primordiale. Les voies de remédiation
non-médicamenteuses qui pourraient reposer sur un remaniement de
l’épigénome – ce qui reste à démontrer – sont à considérer très
sérieusement, avec l’enjeu des moyens humains et d’infrastructure
qu’elles supposent de déployer. Dans tous les cas, la mise en œuvre de
ces voies de remédiation et de suivi des patients implique de pouvoir
poser un diagnostic précoce pour lequel l’utilisation de combinaisons de
biomarqueurs, dont ceux issus de l’épigénétique, pourrait être
déterminante.
Références
[1] Al-Shaer AE, Flentke GR, Berres ME, et al . Exon level machine learning analyses
elucidate novel candidate miRNA targets in an avian model of
fetal alcohol spectrum disorder.
PLoS Comput Biol. 2019;
15: e1006937p.

[2] Andreu-Fernández V, Bastons-Compta A, Navarro-Tapia E, et al . Serum concentrations of IGF-I/IGF-II as
biomarkers of alcohol damage during foetal development and
diagnostic markers of foetal alcohol
syndrome.
Sci Rep. 2019;
9: 1562p.
[3] Aran A, Rosenfeld N, Jaron R, et al . Loss of function of PCDH12 underlies
recessive microcephaly mimicking intrauterine
infection.
Neurology. 2016;
86:2016
-24
[4] Balaraman S, Schafer JJ, Tseng AM, et al . Plasma miRNA profiles in pregnant women
predict infant outcomes following prenatal alcohol
exposure.
PLoS One. 2016;
11: e0165081p.
[5] Beeler E, Nobile ZL, Homanics GE. Paternal preconception every-other-day
ethanol drinking alters behavior and ethanol consumption in
offspring.
Brain Sci. 2019;
9: 56p.
[6] Berg PW, Shaffer J, Vliegenthart ADB, et al . Attending a social event and consuming
alcohol is associated with changes in serum microRNA : a
before and after study in healthy adults.
Biomarkers. 2018;
23:781
-6
[7] Bird A. Perceptions of
epigenetics.
Nature. 2007;
447:396
-8
[8] Bourgeron T. From the genetic architecture to synaptic
plasticity in autism spectrum disorder.
Nat Rev Neurosci. 2015;
16:551
-63
[9] Briggs JA, Wolvetang EJ, Mattick JS, et al . Mechanisms of long non-coding rnas in
mammalian nervous system development, plasticity, disease,
and evolution.
Neuron. 2015;
88:861
-77
[10] Buenrostro JD, Giresi PG, Zaba LC, et al . Transposition of native chromatin for fast
and sensitive epigenomic profiling of open chromatin,
DNA-binding proteins and nucleosome
position.
Nat Methods. 2013;
10:1213
-8
[11] Chabenne A, Moon C, Ojo C, et al . Biomarkers in fetal alcohol
syndrome.
Biomark Genom Med. 2014;
6:12
-22
[12] Chang RC, Wang H, Bedi Y, et al . Preconception paternal alcohol exposure
exerts sex-specific effects on offspring growth and
long-term metabolic programming.
Epigenetics Chromatin. 2019;
12: 9p.
[13] Chang RC, Skiles WM, Chronister SS, et al . DNA methylation-independent growth
restriction and altered developmental programming in a mouse
model of preconception male alcohol
exposure.
Epigenetics. 2017;
12:841
-53
[14] Chater-Diehl EJ, Laufer BI, Singh SM. Changes to histone modifications following
prenatal alcohol exposure : an emerging
picture.
Alcohol. 2017;
60:41
-52
[15] Chater-Diehl EJ, Laufer BI, Castellani CA, et al . Alteration of gene expression, DNA
methylation, and histone methylation in free radical
scavenging networks in adult mouse hippocampus following
fetal alcohol exposure.
PLoS One. 2016;
11: e0154836p.
[16] Chen J, Hutchison KE, Bryan AD, et al . Opposite epigenetic associations with alcohol
use and exercise intervention.
Front Psychiatry. 2018;
9: 594p.
[17] Chen X, Shen Y, Draper W, et al . ATAC-see reveals the accessible genome by
transposase-mediated imaging and
sequencing.
Nat Methods. 2016;
13:1013
-20
[18] Clarke ME, Gibbard WB. Overview of fetal alcohol spectrum disorders
for mental health professionals.
Can Child Adolesc Psychiatr Rev. 2003;
12:57
-63
[19] Clark SL, Costin BN, Chan RF, et al . A whole methylome study of ethanol exposure
in brain and blood : an exploration of the utility of
peripheral blood as proxy tissue for brain in alcohol
methylation studies.
Alcohol Clin Exp Res. 2018;
42:2360
-8
[20] Cobben JM, Krzyzewska IM, Venema A, et al . DNA methylation abundantly associates with
fetal alcohol spectrum disorder and its
subphenotypes.
Epigenomics. 2019;
11:767
-85
[21]CONVERGE consortium. Sparse whole-genome sequencing identifies two
loci for major depressive disorder.
Nature. 2015;
523:588
-91
[22] Davies W, Isles AR, Humby T, et al . What are imprinted genes doing in the
brain?.
Adv Exp Med Biol. 2008;
626:62
-70
[23] Dong E, Guidotti A, Zhang H, et al . Prenatal stress leads to chromatin and
synaptic remodeling and excessive alcohol intake comorbid
with anxiety-like behaviors in adult
offspring.
Neuropharmacology. 2018;
140:76
-85
[24] Drissi I, Deschamps C, Fouquet G, et al . Memory and plasticity impairment after binge
drinking in adolescent rat hippocampus : GluN2A/GluN2B NMDA
receptor subunits imbalance through
HDAC2.
Addict Biol. 2020;
25: e12760p.
[25] Dulac C. Brain function and chromatin
plasticity.
Nature. 2010;
465:728
-35
[26] El Hajj N, Dittrich M, Böck J, et al . Epigenetic dysregulation in the developing
Down syndrome cortex.
Epigenetics. 2016;
11:563
-78
[27] Fransquet PD, Hutchinson D, Olsson CA, et al . Perinatal maternal alcohol consumption and
methylation of the dopamine receptor DRD4 in the offspring :
the triple B study.
Environ Epigenet. 2016;
2: dvw023p.
[28] Gräff J, Kim D, Dobbin MM, et al . Epigenetic regulation of gene expression in
physiological and pathological brain
processes.
Physiol Rev. 2011;
91:603
-49
[29] Guerri C, Bazinet A, Riley EP. Foetal alcohol spectrum disorders and
alterations in brain and behaviour.
Alcohol Alcohol. 2009;
44:108
-14
[30] Guintivano J, Kaminsky ZA. Role of epigenetic factors in the development
of mental illness throughout life.
Neurosci Res. 2016;
102:56
-66
[31] Hagerty SL, YorkWilliams SL, Bidwell LC, et al . DRD2 methylation is associated with executive
control network connectivity and severity of alcohol
problems among a sample of polysubstance
users.
Addict Biol. 2020;
25: e12684p.
[32] Hashimoto-Torii K, Torii M, Fujimoto M, et al . Roles of heat shock factor 1 in neuronal
response to fetal environmental risks and its relevance to
brain disorders.
Neuron. 2014;
82:560
-72
[33] He Y, Ecker JR. Non-CG methylation in the human
genome.
Ann Rev Genomics Hum Genet. 2015;
16:55
-77
[34] Hill SY, Sharma VK. DRD2 methylation and regional grey matter
volumes in young adult offspring from families at ultra-high
risk for alcohol dependence.
Psychiatry Res Neuroimaging. 2019;
286:31
-8
[35] Holtzman L, Gersbach CA. Editing the epigenome : reshaping the genomic
landscape.
Ann Rev Genomics Hum Genet. 2018;
19:43
-71
[36] Hutson JR, Stade B, Lehotay DC, et al . Folic acid transport to the human fetus is
decreased in pregnancies with chronic alcohol
exposure.
PLoS One. 2012;
7: e38057p.
[37]Inserm. Alcool : effets sur la santé.
Collection
Expertise collective. Paris:Éditions Inserm;
2001;
358 pp.
[38] Ishii S, Torii M, Son AI, et al . Variations in brain defects result from
cellular mosaicism in the activation of heat shock
signalling.
Nat Commun. 2017;
8: 15157p.
[39] Ishii S, Hashimoto-Torii K. Impact of prenatal environmental stress on
cortical development.
Front Cell Neurosci. 2015;
9: 207p.
[40] Jarmasz JS, Stirton H, Basalah D, et al . Global DNA methylation and histone
posttranslational modifications in human and nonhuman
primate brain in association with prenatal alcohol
exposure.
Alcohol Clin Exp Res. 2019;
43:1145
-62
[41] Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in
early infancy.
Lancet. 1973;
302:999
-1001
[42] Joubert BR, Håberg SE, Nilsen RM, et al . 450K epigenome-wide scan identifies
differential DNA methylation in newborns related to maternal
smoking during pregnancy.
Environ Health Perspect. 2012;
120:1425
-31
[43] Khalid O, Kim JJ, Kim HS, et al . Gene expression signatures affected by
alcohol-induced DNA methylomic deregulation in human
embryonic stem cells.
Stem Cell Res. 2014;
12:791
-806
[44] Kleiber ML, Diehl EJ, Laufer BI, et al . Long-term genomic and epigenomic
dysregulation as a consequence of prenatal alcohol
exposure : a model for fetal alcohol spectrum
disorders.
Front Genet. 2014;
5: 161p.
[45] Kleiber ML, Mantha K, Stringer RL, et al . Neurodevelopmental alcohol exposure elicits
long-term changes to gene expression that alter distinct
molecular pathways dependent on timing of
exposure.
J Neurodev Disord. 2013;
5: 6p.
[46] Kodituwakku PW. Defining the behavioral phenotype in children
with fetal alcohol spectrum disorders : a
review.
Neurosci Biobehav Rev. 2007;
31:192
-201
[47] Koller G, Zill P, Soyka M, et al . Short-term changes in global methylation and
hydroxymethylation during alcohol
detoxification.
Eur Neuropsychopharmacol. 2019;
29:897
-903
[48] Ladd-Acosta C, Shu C, Lee BK, et al . Presence of an epigenetic signature of
prenatal cigarette smoke exposure in
childhood.
Environ Res. 2016;
144:139
-48
[49] LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in
human neurodevelopmental disorders.
Epigenomics. 2015;
7:1213
-28
[50] LaSalle JM, Powell WT, Yasui DH. Epigenetic layers and players underlying
neurodevelopment.
Trends Neurosci. 2013;
36:460
-70
[51] Laufer BI, Chater-Diehl EJ, Kapalanga J, et al . Long-term alterations to DNA methylation as a
biomarker of prenatal alcohol exposure : from mouse models
to human children with fetal alcohol spectrum
disorders.
Alcohol. 2017;
60:67
-75
[52] Laufer BI, Kapalanga J, Castellani CA, et al . Associative DNA methylation changes in
children with prenatal alcohol exposure.
Epigenomics. 2015;
7:1259
-74
[53] Laufer BI, Mantha K, Kleiber ML, et al . Long-lasting alterations to DNA methylation
and ncRNAs could underlie the effects of fetal alcohol
exposure in mice.
Dis Model Mech. 2013;
6:977
-92
[54] Lecuyer M, Laquerrière A, Bekri S, et al . PLGF, a placental marker of fetal brain
defects after in utero alcohol exposure.
Acta Neuropathol Commun. 2017;
5: 936p.
[55] Lee DS, Luo C, Zhou J, et al . Simultaneous profiling of 3D genome structure
and DNA methylation in single human
cells.
Nat Methods. 2019;
16:999
-1006
[56] Lee S, Rivier C. Gender differences in the effect of prenatal
alcohol exposure on the hypothalamic-pituitary-adrenal axis
response to immune signals.
Psychoneuroendocrinology. 1996;
21:145
-55
[57] Lemoine P, Harousseau H, Borteyru J. Les enfants de parents
alcooliques.
Ouest Med. 1968;
61:476
-482
[58] Lister R, Mukamel EA, Nery JR, et al . Global epigenomic reconfiguration during
mammalian brain development.
Science. 2013;
341: 1237905p.
[59] Liu J, Moyon S, Hernandez M, et al . Epigenetic control of oligodendrocyte
development : adding new players to old
keepers.
Current Opin Neurobiol. 2016a;
39:133
-8
[60] Liu M, Jiang Y, Wedow R, et al . Association studies of up to 1.2 million
individuals yield new insights into the genetic etiology of
tobacco and alcohol use.
Nat Genet. 2019;
51:237
-44
[61] Liu XS, Wu H, Krzisch M, et al . Rescue of fragile X syndrome neurons by DNA
methylation editing of the FMR1 gene.
Cell. 2018;
172:979
-992.e6
[62] Liu XS, Wu H, Ji X, et al . Editing DNA methylation in the mammalian
genome.
Cell. 2016b;
167:233
-247.e17
[63] Loke YJ, Muggli E, Nguyen L, et al . Time- and sex-dependent associations between
prenatal alcohol exposure and placental global DNA
methylation.
Epigenomics. 2018;
10:981
-91
[64] Lucia D, Burgess D, Cullen CL, et al . Periconceptional maternal alcohol consumption
leads to behavioural changes in adult and aged offspring and
alters the expression of hippocampal genes associated with
learning and memory and regulators of the
epigenome.
Behav Brain Res. 2019;
362:249
-57
[65] Luo C, Lancaster MA, Castanon R, et al . Cerebral organoids recapitulate epigenomic
signatures of the human fetal brain.
Cell Rep. 2016;
17:3369
-84
[66] Lussier AA, Morin AM, MacIsaac JL, et al . DNA methylation as a predictor of fetal
alcohol spectrum disorder.
Clin Epigenetics. 2018a;
10: 5p.
[67] Lussier AA, Bodnar TS, Mingay M, et al . Prenatal alcohol exposure : profiling
developmental dna methylation patterns in central and
peripheral tissues.
Front Genet. 2018b;
9: 610p.
[68] Lussier AA, Weinberg J, Kobor MS. Epigenetics studies of fetal alcohol spectrum
disorder : where are we now?.
Epigenomics. 2017;
9:291
-311
[69] Mattson SN, Crocker N, Nguyen TT. Fetal alcohol spectrum disorders :
neuropsychological and behavioral
features.
Neuropsychol Rev. 2011;
21:81
-101
[70] Miranda RC. MicroRNAs and fetal brain development :
implications for ethanol teratology during the second
trimester period of neurogenesis.
Front Genet. 2012;
3: 77p.
[71] Molumby MJ, Anderson RM, Newbold DJ, et al . g-Protocadherins interact with Neuroligin-1
and negatively regulate dendritic spine
morphogenesis.
Cell Rep. 2017;
18:2702
-14
[72] O’Connor MJ, Paley B. Psychiatric conditions associated with
prenatal alcohol exposure.
Dev Disabil Res Rev. 2009;
15:225
-34
[73] Paolozza A, Treit S, Beaulieu C, et al . Diffusion tensor imaging of white matter and
correlates to eye movement control and psychometric testing
in children with prenatal alcohol
exposure.
Hum Brain Mapp. 2017;
38:444
-56
[74] Paolozza A, Munn R, Munoz DP, et al . Eye movements reveal sexually dimorphic
deficits in children with fetal alcohol spectrum
disorder.
Front Neurosci. 2015;
9: 76p.
[75] Paolozza A, Rasmussen C, Pei J, et al . Deficits in response inhibition correlate
with oculomotor control in children with fetal alcohol
spectrum disorder and prenatal alcohol
exposure.
Behav Brain Res. 2014a;
259:97
-105
[76] Paolozza A, Rasmussen C, Pei J, et al . Working memory and visuospatial deficits
correlate with oculomotor control in children with fetal
alcohol spectrum disorder.
Behav Brain Res. 2014b;
263:70
-9
[77] Peadon E, Rhys-Jones B, Bower C, et al . Systematic review of interventions for
children with fetal alcohol spectrum
disorders.
BMC Pediatrics. 2009;
9: 35p.
[78] Perrier F, Viallon V, Ambatipudi S, et al . Association of leukocyte DNA methylation
changes with dietary folate and alcohol intake in the EPIC
study.
Clin Epigenetics. 2019;
11: 57p.
[79] Petrenko CLM, Alto ME. Interventions in fetal alcohol spectrum
disorders : An international perspective.
Eur J Med Genet. 2017;
60:79
-91
[80] Philibert R, Dogan M, Noel A, et al . Genome-wide and digital polymerase chain
reaction epigenetic assessments of alcohol
consumption.
Am J Med Genet Neuropsychiatr
Genet. 2018;
177:479
-88
[81] Pollen AA, Nowakowski TJ, Shuga J, et al . Low-coverage single-cell mRNA sequencing
reveals cellular heterogeneity and activated signaling
pathways in developing cerebral cortex.
Nat Biotechnol. 2014;
32:1053
-8
[82] Popova S, Lange S, Burd L, et al . Health care burden and cost associated with
fetal alcohol syndrome : based on official Canadian
data.
PLoS One. 2012;
7: e43024p.
[83] Portales-Casamar E, Lussier AA, Jones MJ, et al . DNA methylation signature of human fetal
alcohol spectrum disorder.
Epigenetics Chromatin. 2016;
9: 25p.
[84] Rudra P, Shi WJ, Russell P, et al . Predictive modeling of miRNA-mediated
predisposition to alcohol-related phenotypes in
mouse.
BMC Genomics. 2018;
19: 281p.
[85] Schang AL, Sabéran-Djoneidi D, Mezger V. The impact of epigenomic next-generation
sequencing approaches on our understanding of
neuropsychiatric disorders.
Clin Genet. 2018;
93:467
-80
[86] Schultz MD, He Y, Whitaker JW, et al . Human body epigenome maps reveal noncanonical
DNA methylation variation.
Nature. 2015;
523:212
-6
[87] Sharma RP, Grayson DR, Guidotti A, et al . Chromatin, DNA methylation and neuron gene
regulation-the purpose of the package.
J Psychiatr Neurosci. 2005;
30:257
-63
[88] Shema E, Jones D, Shoresh N, et al . Single-molecule decoding of combinatorially
modified nucleosomes.
Science. 2016;
352:717
-21
[89] Sowell ER, Mattson SN, Thompson PM, et al . Mapping callosal morphology and cognitive
correlates: effects of heavy prenatal alcohol
exposure.
Neurology. 2001;
57:235
-44
[90] Streissguth AP, O’Malley K. Neuropsychiatric implications and long-term
consequences of fetal alcohol spectrum
disorders.
Semin Clin Neuropsychiatry. 2000;
5:177
-90
[91] Subbanna S, Joshi V, Basavarajappa BS. Activity-dependent signaling and epigenetic
abnormalities in mice exposed to postnatal
ethanol.
Neuroscience. 2018a;
392:230
-40
[92] Subbanna S, Nagre NN, Shivakumar M, et al . CB1R-mediated activation of caspase-3 causes
epigenetic and neurobehavioral abnormalities in postnatal
ethanol-exposed mice.
Front Mol Neurosci. 2018b;
11: 45p.
[93] Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor
during synaptogenesis prevents postnatal ethanol-induced LTP
deficits and neurobehavioral abnormalities in adult
mice.
Exp Neurol. 2014;
261:34
-43
[94] Subbanna S, Shivakumar M, Umapathy NS, et al . G9a-mediated histone methylation regulates
ethanol-induced neurodegeneration in the neonatal mouse
brain.
Neurobiol Dis. 2013;
54:475
-85
[95] Su Y, Shin J, Zhong C, et al . Neuronal activity modifies the chromatin
accessibility landscape in the adult
brain.
Nat Neurosci. 2017;
20:476
-83
[96] Thompson BL, Levitt P, Stanwood GD. Prenatal exposure to drugs: effects on brain
development and implications for policy and
education.
Nat Rev Neurosci. 2009;
10:303
-12
[97] Toyoda S, Kawaguchi M, Kobayashi T, et al . Developmental epigenetic modification
regulates stochastic expression of clustered protocadherin
genes, generating single neuron
diversity.
Neuron. 2014;
82:94
-108
[98] Tseng AM, Chung DD, Pinson MR, et al . Ethanol exposure increases mir-140 in
extracellular vesicles : implications for fetal neural stem
cell proliferation and maturation.
Alcohol Clin Exp Res. 2019a;
43:1414
-26
[99] Tseng AM, Mahnke AH, Wells AB, et al . Maternal circulating miRNAs that predict
infant FASD outcomes influence placental
maturation.
Life Sci Alliance. 2019b;
2: e201800252p.
[100] Ureña-Peralta JR, Alfonso-Loeches S, Cuesta-Diaz CM, et al . Deep sequencing and miRNA profiles in
alcohol-induced neuroinflammation and the TLR4 response in
mice cerebral cortex.
Sci Rep. 2018;
8: 15913p.
[101] Varadinova M, Boyadjieva N. Epigenetic mechanisms: a possible link
between autism spectrum disorders and fetal alcohol spectrum
disorders.
Pharmacol Res. 2015;
102:71
-80
[102] Veazey KJ, Wang H, Bedi YS, et al . Disconnect between alcohol-induced
alterations in chromatin structure and gene transcription in
a mouse embryonic stem cell model of
exposure.
Alcohol. 2017;
60:121
-33
[103] Veazey KJ, Parnell SE, Miranda RC, et al . Dose-dependent alcohol-induced alterations in
chromatin structure persist beyond the window of exposure
and correlate with fetal alcohol syndrome birth
defects.
Epigenetics Chromatin. 2015;
8: 39p.
[104] Wang SE, Jiang YH. Potential of Epigenetic therapy for
Prader-Willi syndrome.
Trends Pharmacol Sci. 2019;
40:605
-8
[105] Weinberg J, Sliwowska JH, Lan N, et al . Prenatal alcohol exposure: foetal
programming, the hypothalamic-pituitary-adrenal axis and sex
differences in outcome.
J Neuroendocrinol. 2008;
20:470
-88
[106] Wozniak JR, Fuglestad AJ, Eckerle JK, et al . Choline supplementation in children with
fetal alcohol spectrum disorders : a randomized,
double-blind, placebo-controlled trial.
Am J Clin Nutr. 2015;
102:1113
-25
[107] Xu W, Liyanage VRB, MacAulay A, et al . Genome-wide transcriptome landscape of
embryonic brain-derived neural stem cells exposed to alcohol
with strain-specific cross-examination in BL6 and CD1
mice.
Sci Rep. 2019;
9: 206p.
[108] Yi R, Fuchs E. MicroRNAs and their roles in mammalian stem
cells.
J Cell Sci. 2011;
124:1775
-83
[109] Zhang C, Paolozza A, Tseng PH, et al . Detection of children/youth with fetal
alcohol spectrum disorder through eye movement,
psychometric, and neuroimaging data.
Front Neurol. 2019;
10: 80p.
[110] Zhang H, Gelernter J. Review : DNA methylation and alcohol use
disorders. Progress and challenges.
Am J Addict. 2017;
26:502
-15
[111] Zhang Y, Wilson R, Heiss J, et al . DNA methylation signatures in peripheral
blood strongly predict all-cause
mortality.
Nat Commun. 2017;
8: 14617p.
→ Aller vers SYNTHESE