Connaissances fondamentales
2007
1-
Caractéristiques neuropathologiques
La maladie d'Alzheimer provoque des modifications de l'intellect, des comportements et des affects mais il ne s'agit pas d'une maladie psychiatrique. Elle est la conséquence de changements structuraux du cerveau que l'on peut observer à l'œil nu et au microscope, à l'examen post mortem. Il s'agit donc d'une maladie neurologique. L'étude des modifications cérébrales relève d'une spécialité médicale, la neuropathologie (mot traduit de l'anglais et qui signifie anatomo-pathologie neurologique).
La neuropathologie joue un rôle essentiel dans le diagnostic – qui n'est aujourd'hui considéré comme certain que si les constatations microscopiques sont effectivement celles de la maladie d'Alzheimer (McKhann et coll., 1984

) – de même que dans l'interface avec la recherche visant à aboutir à un traitement. Les lésions observées à l'examen neuropathologique sont non seulement nécessaires au diagnostic, mais sont aussi les témoins des mécanismes physiopathologiques. C'est la raison pour laquelle les modèles animaux de la maladie d'Alzheimer ont été développés afin de reproduire ces lésions que la recherche thérapeutique cherche à prévenir ou à guérir.
Macroscopie
Les travaux de neuropathologie montrent que le volume du cerveau est diminué au cours de la maladie d'Alzheimer. Cette atrophie n'est cependant pas homogène. Elle concerne principalement la région hippocampique, l'amygdale temporale et le pôle du lobe temporal (pour revue voir Duyckaerts et Dickson, 2003
). Le poids du cerveau diminue lui aussi de façon hétérogène. Au décès, le poids du lobe temporal chez un patient atteint de maladie d'Alzheimer est en moyenne de 41 % plus léger, le lobe pariétal de 30 % et le lobe frontal de 14 % que chez une personne de même âge exempte de maladie neurologique (Najlerahim et Bowen, 1989
). En revanche, le poids du lobe occipital est généralement similaire à celui des sujets témoins.
L'atrophie du cortex ne porte pas seulement sur la substance grise. Elle intéresse également la substance blanche (où passent les axones et les dendrites des neurones situés dans la substance grise). Le cortex tel qu'il peut être vu en coupe apparaît comme un long ruban, replié sur lui-même en circonvolutions. On peut mesurer la longueur de ce ruban et son épaisseur. Alors que l'épaisseur est diminuée dans la maladie d'Alzheimer, c'est surtout la longueur du ruban cortical qui est réduite (Duyckaerts et coll., 1985
), ce qui suggère que la perte de neurones et de connexions se produit en colonnes perpendiculaires à la surface du cortex. La perte de volume du cortex est associée à une dilatation des ventricules, qui reste souvent modérée. C'est ainsi qu'on a évalué à 57 % le taux de patients atteints de maladie d'Alzheimer et présentant une dilatation ventriculaire à l'examen neuropathologique (Hubbard et Anderson, 1981
). On peut ainsi comprendre que la dilatation ventriculaire observée au scanner ait une mauvaise sensibilité (46±20 %) mais une relativement bonne spécificité (90±7 %) vis-à-vis de la maladie d'Alzheimer (DeCarli et coll., 1990
).
À cet égard, aucune des modifications macroscopiques constatées au cours de la maladie d'Alzheimer ne peut être considérée comme spécifique de la pathologie. Seul l'examen microscopique du cerveau permet d'affirmer le diagnostic.
Microscopie
L'examen microscopique est effectué sur des préparations provenant d'échantillons de quelques cm2 de surface, coupés à 5 et jusqu'à 10 mm d'épaisseur pour les microscopes habituels à lumière. Des coupes de quelques mm2 et de quelques dizaines de nm d'épaisseur sont utilisées en microscopie électronique. Dans les deux cas, les coupes doivent être préparées : pratiquement transparentes à la lumière, elles sont colorées par diverses techniques. L'étude de la maladie d'Alzheimer a été rendue possible par la visualisation des lésions au moyen des techniques argentiques, développées à la fin du XIXe et au début du XXe siècle : elles consistent à faire précipiter sur la coupe microscopique, sous forme d'argent métallique, l'argent ionique dissous dans la solution où est immergée la coupe. Le précipité d'argent métallique se dépose principalement sur les structures fibrillaires normales (en particulier les axones) et anormales (les lésions neurofibrillaires de la maladie d'Alzheimer). Actuellement, on utilise, en microscopie photonique comme en microscopie électronique, l'immunohistochimie, une technique qui recourt à un anticorps dirigé spécifiquement contre la protéine qu'on cherche à mettre en évidence. Le développement de cette méthode a grandement contribué aux connaissances acquises sur les lésions de la maladie d'Alzheimer et sur leur constitution chimique.
Les échantillons doivent être prélevés dans des régions précises ; en effet, les lésions de la maladie d'Alzheimer sont réparties de façon stéréotypée mais très hétérogène dans le cerveau. Si les échantillons étaient collectés au hasard, un grand nombre d'entre eux apparaîtraient normaux à l'examen même très détaillé. C'est la raison pour laquelle, dans les paragraphes suivants, il ne suffit pas d'exposer la nature des lésions ; il faut aussi préciser leur topographie et comprendre la séquence de leur apparition.
Nature des lésions
On peut classer les lésions en deux groupes : celles qui consistent en modifications positives (les deux principales sont les plaques séniles et les dégénérescences neurofibrillaires) et celles qui sont en rapport avec des modifications négatives (perte de neurones et de synapses). Ce sont les lésions positives qui permettent le diagnostic ; les lésions négatives ont une grande importance physiopathologique mais elles sont peu spécifiques et souvent difficiles à affirmer. On peut aussi classer les lésions selon les protéines qui s'y accumulent : peptide β-amyloïde (Aβ) ou protéine Tau ; c'est l'option que nous avons choisie ici. Nous verrons qu'il n'y a pas superposition exacte entre la classification biochimique (accumulation de peptide Aβou de protéine Tau) et la classification morphologique (plaque sénile et dégénérescence neurofibrillaire) parce que la plaque sénile, composite, comporte les deux protéines.
Lésions positives
Les deux lésions positives sont l'accumulation extracellulaire de peptide Aβet l'accumulation intracellulaire de protéine Tau.
Accumulation extracellulaire de peptide Aβ
Le peptide A4 (Glenner et Wong, 1984
), amyloïde ou Aβ(comme il est habituel de l'appeler aujourd'hui), est naturellement produit dans le cerveau (Seubert et coll., 1992
). Il est issu, à la suite de deux coupures enzymatiques (par les β- et ɣ- secrétases), d'une protéine transmembranaire, la βAPP (pour
β-Amyloid Precursor Protein, Kang et coll., 1987
). Dans la maladie d'Alzheimer, pour des raisons qui restent encore mal comprises, le peptide s'accumule principalement en dehors des cellules et échappe aux divers systèmes biologiques d'élimination, enzymatiques ou cellulaires. Des accumulations intracellulaires de peptide Aβont été rapportées chez la souris transgénique (APP et préséniline 1 mutées) (Langui et coll., 2004
) et, plus rarement, chez l'homme (Gouras et coll., 2000
). Des observations récentes laissent penser que la présence de peptide Aβintracellulaire n'est pas liée à la maladie (Wegiel et coll., 2007
). L'agrégation provient d'une modification des propriétés stucturales du peptide : il s'enrichit en feuillets β-plissés et, devenu insoluble, précipite dans le tissu nerveux. Ce mécanisme n'est pas propre au peptide Aβ. Il s'observe dans d'autres affections, neurologiques ou non. Les protéines enrichies en feuillets β-plissés qui ont précipité dans le milieu extracellulaire sont qualifiées d'« amyloïdes » (Glenner, 1980
). Elles ont des propriétés communes : elles résistent aux procédés habituels de solubilisation, sont mises en évidence par divers colorants comme le rouge Congo ou la thioflavine et apparaissent fibrillaires en microscopie électronique.
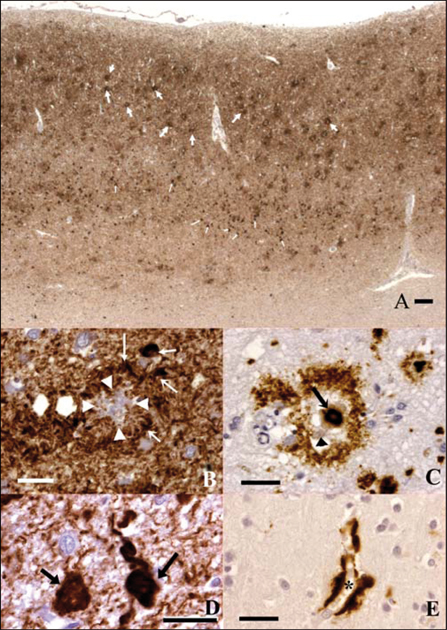
Le peptide Aβs'accumule sous diverses formes (Delaère et coll., 1991
) : le « dépôt focal » constitue le cœ ur, extracellulaire et sphérique, de la plaque sénile (figure 1.1C
). Il est amyloïde et en particulier coloré par le rouge Congo (Divry, 1927
). La plaque sénile comporte une couronne faite de prolongements nerveux. Des données récentes obtenues par la spectrométrie de masse laissent penser que le cœ ur de la plaque sénile n'est constitué que de peptide Aβ(Soderberg et coll., 2006
).
Le « dépôt diffus » n'est pas amyloïde (Tagliavini et coll., 1988
) et peut seulement être visualisé par immunohistochimie utilisant des anticorps dirigés contre le peptide Aβ. Il est de plus grande taille (pouvant atteindre quelques centaines de microns), moins dense et souvent, du point de vue morphologique, mal limité. Le dépôt diffus n'a pas de composante nerveuse mais il peut modifier la géométrie des dendrites ou des axones qui le traversent ou s'y trouvent (Knowles et coll., 1999
; Buldyrev et coll., 2000
). Les dépôts diffus peuvent être présents en grand nombre chez des personnes âgées dépourvues de troubles intellectuels, ou bien marqués (Delaère et coll., 1990
; Dickson et coll., 1991
), une condition qui a été dénommée « vieillissement pathologique » (Dickson, et coll., 1991
). On peut aussi penser que le dépôt diffus constitue le stade précoce de la plaque sénile en voie de constitution : ce pourrait en effet être le cas dans le cortex cérébral mais dans certaines régions cérébrales, les dépôts sont toujours diffus et ne constituent jamais de plaques séniles typiques comportant un cœ ur d'amyloïde et une couronne nerveuse. C'est le cas, par exemple, dans le striatum (Brilliant et coll., 1997
) ou le cervelet (Joachim et coll., 1989
). Le dépôt « laineux » (
fleecy) qui siège dans les couches profondes du cortex entorhinal (Thal et coll., 1999
) et le dépôt en flaque (
lake-like) qui intéresse le présubiculum (Wisniewski et coll., 1998
) sont volumineux et concernent une ou plusieurs couches corticales sans interruption (c'est-à-dire sans formation de plaques sphériques). Le dépôt peut, au contraire, être dense et de petite taille (moins de 5 mm) : c'est le dépôt stellaire (Delaère et coll., 1991
). Le dépôt de peptide Aβse produit volontiers en superficie du cortex (dépôt sous-pial). Il peut prendre une forme particulière (dite « en laine de coton »,
cotton wool) : ces plaques ne prennent pas ou peu les colorants des substances amyloïdes, sont bien visibles après coloration par l'hématéine-éosine, et sont marquées par les anticorps anti-Aβ 42. Elles ont été initialement décrites au cours de la maladie d'Alzheimer familiale due à une délétion du gène de la préséniline 1 (Crook et coll., 1998
). Plus récemment, elles ont aussi été signalées dans des cas sporadiques de maladie d'Alzheimer, mais alors presque toujours associées à des plaques séniles typiques (Le et coll., 2001
).
Le peptide Aβpeut aussi s'accumuler dans la paroi des vaisseaux, souvent de façon segmentaire (seule une partie du vaisseau est touchée) ou même sur une partie de sa circonférence. Les petites artères perforantes du cortex cérébral sont le plus souvent intéressées mais des artères méningées plus volumineuses, dans certains cas des capillaires, et peut-être des veines peuvent aussi être affectés. Le terme de « dyshorique » dû à Morel (Morel, 1950
) s'applique à un dépôt vasculaire qui semble sortir de la paroi pour s'étendre dans le parenchyme (figure 1.1E
), une topographie qui suggérait à cet auteur un trouble (dys-) de la barrière (horos) hémato-encéphalique. Il désigne aujourd'hui l'accumulation de peptide Aβ, dans la paroi de capillaires ou de petites artères, entourée de prolongements neuritiques. Le terme d'angiopathie « congophile » désigne au contraire l'atteinte des gros vaisseaux. Les termes de micro- et de macro-angiopathie sont aussi utilisés pour décrire l'atteinte des petites ou des grosses artères. L'angiopathie amyloïde est, sinon constante, au moins très fréquente dans la maladie d'Alzheimer sporadique (Joachim et coll., 1987
et 1988
). L'angiopathie amyloïde des capillaires est statistiquement liée à la sévérité des lésions neuritiques de la maladie, alors que la macroangiopathie ne l'est pas (Attems et Jellinger, 2004
).
L'angiopathie amyloïde est particulièrement sévère dans les cas associés à certaines mutations de l'APP ou de la préséniline 1. C'est ainsi que la maladie vasculaire est au premier plan (Rozemuller et coll., 1993
) dans la mutation dite hollandaise qui remplace la glutamine par l'acide glutamique en position 693 de l'APP (Levy et coll., 1990
). La présence de deux allèles de l'apolipoprotéine E4 (Apoe4) est, elle aussi, associée à une angiopathie marquée (Vidal et coll., 2000
) ; des vaisseaux amyloïdes sont fréquemment observés au centre des plaques séniles. Ces plaques séniles « angiocentriques » sont aussi fréquentes dans la mutation « flamande » de l'APP (Kumar-Singh et coll., 2002
).
En réalité, parler de peptide Aβcomme s'il s'agissait d'un peptide unique est une généralisation. On sait aujourd'hui qu'il existe une hétérogénéité carboxy-terminale. Le peptide se termine parfois à l'acide aminé 42 (Aβ 42), parfois à l'acide aminé 40 (Aβ). Les dépôts diffus sont principalement constitués de peptide Aβ 42, les dépôts vasculaires comportent principalement de l'Aβ 40 (Suzuki et coll., 1994
), et le cœ ur des plaques séniles comporte les deux formes du peptide. D'autre part, il arrive fréquemment que le peptide ne commence pas à l'acide aminé 1 parce que sa partie amino-terminale a été tronquée. Ces formes N-tronquées sont hétérogènes et abondantes dans les dépôts diffus (Tekirian et coll., 1998
; Thal et coll., 1999
; Russo et coll., 2001
; Liu et coll., 2006
).
Le peptide Aβen solution forme des oligomères, qui pourraient avoir une grande importance physiopathologique (Walsh et coll., 2002
). Ils provoquent une inhibition fonctionnelle de la potentialisation à long terme observée sur des tranches d'hippocampe en culture (Walsh et coll., 2002
; Wang et coll., 2004
) et se fixent aux synapses
in vitro, provoquant des modifications des épines dendritiques (Lacor et coll., 2007
).
Accumulation intracellulaire de protéine Tau
Les colorations argentiques furent les premières à permettre de visualiser la pathologie neurofibrillaire. Aloïs Alzheimer utilisa la technique de Bielschowsky (Bielschowsky, 1902
et 1903
), décrite peu de temps avant sa communication princeps (Alzheimer, 1907
). Avec cette méthode, l'accumulation de matériel fibrillaire dans le corps cellulaire du neurone est soulignée par le dépôt d'argent métallique, « argyrophilie » mise à profit dans de nombreuses autres techniques (comme celles de Bodian et de Gallyas, encore couramment utilisées aujourd'hui). L'adjectif neurofibrillaire qui est utilisé ici est dû, en réalité, à une erreur d'interprétation. Les « neurofibrilles » sont des constituants normaux des neurones identifiés dès la fin du XX
e siècle. Les progrès de la microscopie électronique et de la biologie cellulaire ont montré qu'il s'agissait de « neurofilaments », un des éléments du cytosquelette, constitué de protéines spécifiques. Mais la
Fibrillenveränderung d'Alzheimer traduite en français par « dégénérescence neurofibrillaire » (DNF) et en anglais par «
neurofibrillary tangle » n'est pas constituée de neurofilaments, comme on l'a d'abord cru (Anderton et coll., 1982
) mais d'une protéine normale qui intervient dans la polymérisation des neurotubules, la protéine Tau (Brion et coll., 1985
; Grundke-Iqbal et coll., 1986
). Cette protéine a été initialement isolée par des spécialistes des tubules (Cleveland et coll., 1977
) ; son séquençage a été réalisé bien plus tardivement (Goedert et coll., 1988
et 1989
). Six isoformes sont connues. La protéine comporte des segments répétitifs au nombre de 3 (Tau 3R) ou de 4 (Tau 4R). Les lésions de la maladie d'Alzheimer comportent à la fois des formes 3R et 4R, les unes et les autres anormalement phosphorylées.
Au cours de la maladie d'Alzheimer, la protéine Tau s'accumule presque exclusivement dans le neurone dont elle occupe les différents compartiments. Dans le corps cellulaire, les amas de protéine Tau constituent la dégénérescence neurofibrillaire ; ils sont aussi présents dans les extrémités axonales qui entourent le cœ ur de la plaque sénile et forment sa « couronne » ; les « filaments du neuropile » (
neuropil threads) qu'on appelle aussi en français, les « fibres tortueuses » (Duyckaerts et coll., 1989a
), sont principalement en rapport avec l'accumulation de protéine Tau dans les dendrites (Braak et coll., 1986
; Braak et Braak, 1988
). Enfin, la concentration de la protéine Tau peut être élevée dans le cytoplasme du neurone, sans que l'agrégation ne se soit encore produite. Le marquage diffus du corps cellulaire et des prolongements nerveux par les anticorps anti-Tau est désigné par le terme de pré-DNF (Bancher et coll., 1989
).
Plaque sénile : un cœ ur de peptide Aβet une couronne de protéine Tau
La plaque sénile « classique » ou « mûre » (
mature en anglais) est constituée d'un dépôt focal amyloïde de peptide Aβet d'une couronne faite d'un entrelacs de prolongements axonaux chargés de protéine Tau ou, pour certains d'entre eux, de βAPP. Nombre de ces prolongements sont aussi marqués par les anticorps anti-ubiquitine (He et coll., 1993
). Cette plaque sénile classique comporte invariablement une ou plusieurs cellules microgliales activées (Arends et coll., 2000
). Leur rôle a été discuté : pour certains, elles sont indispensables à la transformation amyloïde du peptide Aβ(Frackowiak et coll., 1992
) ; pour d'autres, elles phagocytent les fibrilles d'amyloïde déjà produits. Quoi qu'il en soit, elles expriment des chimiokines, contribuant à entretenir une inflammation locale, à bas bruit, sans composante lymphocytaire. Cette réaction inflammatoire comporte des composants précoces de la cascade du complément (McGeer et coll., 1989
), de l'alpha 1 anti-chymotrypsine (Abraham et coll., 1988
), et des cytokines pro-inflammatoires (Dickson et coll., 1993
). Les expériences de « vaccination » par des préparations comportant des épitopes du peptide Aβadministrés de façon systémique, indiquent que la microglie activée peut éliminer la composante amyloïde de la plaque sénile (Schenk et coll., 1999
). La plaque comporte également du cholestérol (Mori et coll., 2001
) et de l'apolipoprotéine E, son transporteur (Uchihara et coll., 1995
; Dickson et coll., 1997
).
La couronne de la plaque sénile est principalement constituée de prolongements axonaux (Schmidt et coll., 1991
). L'origine de ces axones est encore imparfaitement connue. Certains sont catécholaminergiques (Berger et coll., 1980
), mais beaucoup paraissent issus des connexions cortico-corticales (Duyckaerts et coll., 1986
). Chez la souris transgénique APP (qui développe des plaques séniles), les axones qui entourent les dépôts de peptide Aβproviennent en majorité de connexions cortico-corticales (Delatour et coll., 2003
). La couronne de la plaque sénile se développe après le dépôt de peptide Aβ(Metsaars et coll., 2003
).
Lésions négatives
La perte neuronale et la perte synaptique sont les deux principales lésions négatives.
Perte neuronale : importance et mécanisme
L'ampleur de la perte neuronale est très diversement appréciée, en grande partie du fait de l'utilisation de méthodes variées d'estimation de la densité neuronale (nombre de profils neuronaux par mm
2 de coupe microscopique ou de neurones par mm
3 d'échantillon) et d'évaluation du nombre total de neurones dans le cortex cérébral ou dans une aire corticale déterminée. Le compte non corrigé des profils neuronaux sur une coupe examinée au microscope est biaisé : il surestime en effet les grands neurones (qui ont une probabilité plus grande d'être coupé) par rapport aux petits. Diverses méthodes stéréologiques (visant à reconstituer la densité dans un volume à partir de son estimation sur une coupe) ont été utilisées pour pallier ce biais. La technique dite du « disecteur » (pour « deux sections ») a été la plus utilisée. Elle consiste à ne comptabiliser que les profils neuronaux présents sur l'une des coupes et absents de l'autre – ce qui évite les effets de recoupe à l'origine du biais. L'importance physiopathologique de la mort neuronale a fait l'objet de discussions : pour certains, elle constitue un élément essentiel et précoce de la physiopathologie, à l'origine directe des symptômes (Gomez-Isla et coll., 1996
et 1997
). Pour d'autres, au contraire, elle survient tardivement, à une phase de la maladie où les symptômes sont déjà installés (Regeur et coll., 1994
; Grignon et coll., 1998
). Les premières études n'utilisant pas la méthode du disecteur mentionnaient une perte des grands neurones (Terry et coll., 1981
; Hansen et coll., 1988
), qui, à la lumière de la stéréologie, pouvait être interprétée comme la conséquence tout aussi bien d'une diminution de volume des grands neurones que d'une mort cellulaire (Duyckaerts et coll., 1989b
). Les analyses initiales ayant eu recours à la technique du disecteur ont permis de montrer qu'il n'existait pas de « perte neuronale néocorticale globale » dans la maladie d'Alzheimer (Regeur et coll., 1994
). En revanche, la même méthode a démontré que la perte neuronale était très marquée dans des aires corticales spécifiques (comme l'aire entorhinale) – ou même dans des couches corticales spécifiques – et dépassait le nombre des dégénérescences neurofibrillaires qui pourraient donc ne pas être responsables, ou ne pas être responsables à elles seules, de la mort neuronale (Gomez-Isla et coll., 1996
et 1997
). La méthode des polygones de Voronoï, qui permet d'analyser des variations locales de densité cellulaire, a elle aussi montré que la perte neuronale prédominait dans des couches spécifiques, celles précisément affectées par les DNF (couches III et V) (Grignon, et coll., 1998
). La perte neuronale est statistiquement liée à la densité des DNF, surtout nette dans les échantillons où elles sont plus nombreuses que 5/mm
2. Le nombre total de neurones dans le lobe pariétal était inférieur de 98 millions chez les patients ayant plus de 5 DNF/mm
2. L'analyse multivariée montre que le statut intellectuel est mieux corrélé à l'augmentation de la densité des DNF qu'à la perte neuronale (Grignon et coll., 1998
).
Le mécanisme de la mort neuronale a été discuté. Plusieurs arguments laissent penser qu'elle pourrait être provoquée par les DNF elles-mêmes. La perte neuronale est en effet marquée dans les régions où les DNF sont nombreuses (Grignon et coll., 1998
). Elles peuvent subsister – à l'état de « fantôme » (selon l'expression anglaise de «
ghost tangles ») – lorsque les neurones dont elles occupaient le cytoplasme meurent. De nombreux « fantômes » sont observés dans l'hippocampe ou l'aire entorhinale. La somme « fantômes plus neurones survivants » est proche du nombre total de neurones observés dans les cas normaux (Cras et coll., 1995
). La question d'une apoptose a souvent été soulevée (Cotman et Anderson, 1995
). Rappelons que l'apoptose est une mort cellulaire programmée, nécessitant la synthèse de nouvelles protéines et comportant une fragmentation régulière de l'ADN nucléaire qui, en électrophorèse, migre en bandes disposées en échelle. La technique de marquage
in situ des extrémités polynucléotidiques («
in situ end labelling » ou Isel) a montré que l'ADN était souvent fragmenté dans les neurones au cours de la maladie d'Alzheimer, mais ces résultats ne signifient pas que l'apoptose est fréquente : la migration en échelle n'a pas pu être mise en évidence (Cotman, 1998
), les stigmates morphologiques d'apoptose font défaut (Stadelmann et coll., 1998
) et le nombre de cellules marquées par la technique Isel n'est pas compatible avec une évolution chronique. L'utilisation, plus récente, d'anticorps dirigés contre la caspase-3 activée, une enzyme intervenant dans la chaîne de réactions aboutissant à l'apoptose, a permis de conclure que la densité des neurones apoptotiques était beaucoup plus faible (1 neurone apoptotique pour 1 100 à 5 000 neurones examinés) que la technique Isel ne l'avait laissé croire. Ces valeurs, contrairement à celles qui avaient été avancées auparavant, sont compatibles avec l'évolution chronique de la maladie (Stadelmann et coll., 1999). Une relation entre les marqueurs de l'apoptose et la présence de DNF a été rapportée (Rohn et coll., 2001
). L'expression, par certains neurones en dégénérescence, de protéines impliquées dans la régulation du cycle cellulaire a conduit à formuler l'hypothèse selon laquelle le neurone, normalement post-mitotique, tenterait d'entrer dans une phase de prolifération (Nagy et coll., 1998
). L'impossibilité de se diviser le conduirait à la mort.
Perte synaptique
Le rôle physiopathologique de la perte synaptique a été jugé central par quelques auteurs (Terry et coll., 1991
et 1994
; Masliah et coll., 1993
et 1994
; Masliah et Terry, 1993
). Elle a été appréciée par différentes techniques. La microscopie électronique a révélé une diminution de la densité des synapses mais une augmentation parallèle de leur surface d'apposition, ce qui suggère que la taille des synapses augmente tandis que leur nombre diminue (Scheff et coll., 1990
, 1993
et 1996
; Scheff et Price, 1993
et 2006
).
L'immunohistochimie de la synaptophysine, une protéine vésiculaire présynaptique, révèle une diminution importante de l'immunoréactivité, interprétée comme la conséquence de la perte des synapses (Masliah et coll., 1989
). Cependant, l'appréciation de la perte synaptique varie selon les marqueurs utilisés et apparaît beaucoup plus prononcée pour les protéines liées aux vésicules synaptiques que pour celles qui sont localisées à la membrane (Shimohama et coll., 1997
). La perte synaptique ne semble pas liée à l'accumulation de peptide Aβ(Boncristiano et coll., 2005
) et n'est sans doute pas le meilleur corrélat des troubles cognitifs (Dickson et coll., 1995
). Dans cette dernière étude, c'était à nouveau la densité des DNF qui paraissait la plus étroitement liée à l'état cognitif. Les oligomères de peptide Aβse fixent aux synapses et la possibilité d'une « apoptose synaptique » a été suggérée (Mattson et coll., 1998
).
Topographie des lésions
Les lésions de la maladie d'Alzheimer, pratiquement confinées à la substance grise (c'est-à-dire aux régions contenant les corps cellulaires des neurones) ont une répartition remarquablement hétérogène . Elle est différente pour le peptide Aβet la protéine Tau. Les dépôts, diffus ou focaux, de peptide Aβsont principalement trouvés dans l'isocortex ; des dépôts uniquement diffus sont constatés dans le striatum (Gearing et coll., 1997
), le thalamus (Rub et coll., 2002
) et le cervelet (Mann et coll., 1990
), à des stades plus tardifs. Des dépôts d'Aβsont aussi constatés dans le tronc cérébral (Iseki et coll., 1989
).
Les cellules pyramidales des champs ammoniens (CA) de l'hippocampe, de la couche II du cortex entorhinal et de la couche III de l'isocortex, les cellules multipolaires du noyau basal de Meynert sont celles qui sont le plus souvent et le plus précocement affectées par la pathologie neurofibrillaire. Au contraire, les cellules granulaires du gyrus denté et de la couche IV de l'isocortex ne sont atteintes que rarement et tardivement (figure 1.2).
Chronologie des lésions
L'évolution relative des lésions les unes par rapport aux autres est bien connue (contrairement à la vitesse avec laquelle elles se forment chez un individu donné, qui reste impossible à apprécier aujourd'hui où seul l'examen post mortem permet d'étudier la topographie des lésions). Peu d'études permettent de se faire une idée précise de la prévalence des lésions dans la population générale.
Chronologie d'apparition des lésions les unes par rapport aux autres
Quelle est la lésion initiale, le dépôt de peptide Aβou l'accumulation intracellulaire de protéine Tau ? La réponse est encore discutée : des dépôts de peptide Aβont été observés, en l'absence de DNF, dans le cortex de patients trisomiques 21 décédés à un âge précoce (Mann et Esiri, 1989
) ou chez des patients porteurs de mutations responsables de maladie d'Alzheimer familiale avant l'apparition des premiers symptômes (Troncoso et coll., 1998
). Mais ces constatations sont exceptionnelles. La détection systématique chez un grand nombre de sujets de dépôts de peptide Aβet de protéine Tau en fonction de l'âge a montré que, dans une proportion significative de la population, des DNF étaient présentes en l'absence de dépôts d'Aβ(Braak et Braak, 1997
; Duyckaerts et Hauw, 1997a
) mais conclure à l'absence de dépôts ne peut être aujourd'hui qu'une approximation. Il est en effet impossible d'examiner l'ensemble du cerveau au microscope. L'observation ne peut porter que sur un nombre défini et limité d'échantillons, le résultat étant ensuite extrapolé aux régions non examinées. La précession au moins apparente des DNF sur l'accumulation de peptide Aβdans les formes sporadiques n'est pas prévue par l'hypothèse de la cascade amyloïde (Hardy, 1992
) selon laquelle la perturbation du métabolisme du peptide Aβest à l'origine de l'ensemble des lésions. Selon un modèle alternatif à deux variables, « le vieillissement cérébral » expliquerait l'apparition des DNF, alors que la perturbation du métabolisme du peptide Aβserait propre à la maladie d'Alzheimer. L'interaction entre les DNF et les dépôts d'Aβpermettrait seule l'extension des lésions (Duyckaerts et Hauw, 1997b
).
Une fois constituées, les DNF subsistent longtemps – peut-être pendant des décennies – d'abord dans le neurone, puis, le neurone une fois mort, dans le milieu extracellulaire sous la forme de DNF « fantômes ». Non résorbées par l'organisme, il est possible de les suivre dans les différentes aires corticales où elles apparaissent successivement : d'abord le cortex transentorhinal et entorhinal, puis l'hippocampe (secteurs pyramidaux avec respect au moins relatif du gyrus denté et du présubiculum). Les aires associatives sont ensuite touchées, les aires primaires sont les dernières intéressées (Braak et Braak, 1991
; Delacourte et coll., 1999
; Metsaars, et coll., 2003
). Des informations plus récentes laissent supposer que l'aire 19 (cortex visuel associatif) pourrait être affectée dès les premiers stades de la maladie (McKee et coll., 2006
). Ces débuts focaux dans l'isocortex sont peut-être plus variés et plus fréquents qu'on ne le pense, comme le suggèrent, par exemple, les aphasies progressives primaires en rapport avec la maladie d'Alzheimer. On dispose d'informations beaucoup moins précises sur la chronologie des lésions dans les noyaux sous-corticaux, à l'exception du noyau basal de Meynert (Sassin et coll., 2000
) où des altérations neurofibrillaires sont présentes dès les premiers stades. Le dépôt diffus de peptide Aβest la première des lésions amyloïdes à apparaître. Elle pourrait être précédée ou accompagnée de l'accumulation intracellulaire de peptide Aβ, une lésion bien documentée chez la souris transgénique (Langui et coll., 2004
; Knobloch, 2006
).
Chronologie d'apparition dans la population
Malgré le nombre considérable d'études neuropathologiques concernant la maladie d'Alzheimer, des chiffres solides de prévalence des lésions en population ou chez les patients en long séjour sont remarquablement peu nombreux. Dans l'étude de Braak et Braak (1997
), la sélection de la population n'avait pas fait l'objet d'une procédure particulière ; il est donc probable que les valeurs avancées soient surestimées (car les spécimens provenaient notamment de médecine légale et d'hospices où la proportion de déments est plus élevée que dans la population générale). Dans cette étude, portant sur 2 661 cerveaux, des DNF sont observées dans la moitié de la population à l'âge de 47 ans – voir analyse de ces données dans Duyckaerts et Hauw, 1997b
. La prévalence ne cesse de croître ensuite pour atteindre 100 % chez les centenaires (Hauw et coll., 1986
). Les dépôts de peptide Aβatteignaient une prévalence identique à celle des accumulations de protéine Tau avec un décalage de plusieurs décennies (Braak et Braak, 1997
; Duyckaerts et Hauw, 1997b
). Ils étaient également systématiquement détectés chez les centenaires (Delaère et coll., 1993
).
Corrélations entre la densité des lésions et le déficit intellectuel
De nombreuses études ont cherché à établir la relation entre la densité des différentes lésions et le déficit intellectuel. Nombre d'entre elles ont conclu à l'importance des DNF, ou de façon plus spécifique des lésions Tau positives par rapport aux dépôts de peptide Aβ(Wilcock et Esiri, 1982
; Dickson et coll., 1995
; Duyckaerts et coll., 1995
et 1997
; Berg et coll., 1998
; Giannakopoulos et coll., 2007
). La perte neuronale, plus difficile à évaluer, est généralement moins bien corrélée avec le déficit cognitif (Gomez-Isla, et coll., 1996
; Grignon et coll., 1998
). La baisse de la synaptophysine corticale a été considérée comme le meilleur corrélat de la démence (Terry, et coll., 1994
) mais ce point a lui aussi été contesté (Dickson et coll., 1995
) comme nous l'avons signalé.
Diagnostic neuropathologique de la maladie d'Alzheimer
Les experts qui, sous la signature de Zaven Khachaturian, publièrent les premiers critères formels de maladie d'Alzheimer tentèrent de délimiter une population de cas
post mortem qui pouvaient être utilisés pour la recherche (Khachaturian, 1985
). Ils proposèrent un protocole de prélèvement des échantillons, affirmèrent que la plaque sénile était la lésion la plus significative et donnèrent une valeur seuil à partir de laquelle le diagnostic de maladie d'Alzheimer était porté. Cette démarche binaire de diagnostic ne résista pas à l'expérience : les critères proposés par un consortium de centres Alzheimer nord-américains, comportaient une analyse semi-quantitative de la densité des plaques : « nombreuses », « en nombre modéré », « rares », « absentes » qui aboutissait à des catégories diagnostiques de maladie d'Alzheimer « neuropathologiquement possibles » ou « probables », entre les extrêmes que constituaient le « cerveau normal » et la « maladie d'Alzheimer certaine » (Mirra et coll., 1991
).
On découvrit alors que le compte de plaques séniles pouvait être élevé chez des sujets sans trouble cognitif apparent ou avec des troubles minimes et que le meilleur corrélat de la démence était la pathologie Tau. Ces deux observations conduisirent à de nouveaux critères dits du NIA-Reagan (Institut National du Vieillissement-Institut Reagan) (Ball et coll., 1997
). Ceux-ci ont modifié la perspective, puisqu'ils déterminent la probabilité que les lésions observées
post mortem expliquent la démence dont souffrait le patient. En d'autres termes, le cas de sujets non déments ou dont l'état intellectuel n'est pas connu, n'est pas considéré. La démence est reliée aux lésions avec un « haut degré de probabilité », un « degré intermédiaire » ou un « bas degré », en fonction de la pathologie neurofibrillaire, évaluée par les stades de Braak et Braak (Braak et Braak, 1991
) et la densité des plaques, évaluées de façon semi-quantitative (comme dans les critères du CERAD). Les lésions asymptomatiques ne sont pas prises en compte.
Les données récentes laissent penser qu'un diagnostic binaire est à l'origine de difficultés pratiques, en particulier pour l'utilisation des échantillons par les chercheurs. Les lésions d'Alzheimer n'occupent pas nécessairement des aires contiguës. Un chercheur peut donc recevoir un échantillon provenant d'une aire normale ou peu atteinte provenant d'un cas où le diagnostic de maladie d'Alzheimer a été porté. D'autre part, les lésions d'Alzheimer sont beaucoup plus prévalentes qu'on ne le pensait naguère. Le cortex entorhinal, par exemple, est rarement normal à partir d'un certain âge : le chercheur peut donc recevoir des échantillons anormaux provenant d'un sujet réputé normal. Enfin, les lésions d'Alzheimer peuvent ne constituer qu'un paramètre dans une équation complexe faisant également intervenir les altérations vasculaires ou d'autres lésions neurodégénératives comme celles de la maladie de Parkinson. En conséquence, la recherche ne peut être entreprise que sur un échantillon étiqueté sans lésion ou pourvu de lésions d'Alzheimer et non sur un cas réputé normal (où tous les échantillons seraient normaux – ce qui est erroné) ou réputé « maladie d'Alzheimer » (où tous les échantillons seraient anormaux – ce qui ne l'est pas moins). L'évaluation neuropathologique doit reposer sur une analyse topographique formalisée par les « stades de Braak » qui déterminent les aires corticales où sont localisées les lésions Tau et la « phase de Thal » (2002
) – qui décrivent la topographie des dépôts de peptide Aβ. Dans les aires touchées, la sévérité des lésions détermine leur « grade » (Metsaars et coll., 2003
). Les critères ICDNS (
International Classification of the Diseases of the Nervous System1
; Achim et coll., 2002
) sont fondés sur ces principes.
Questions méthodologiques
Le cadre neuropathologique de la maladie d'Alzheimer est posé depuis une vingtaine d'années. Il serait erroné d'en conclure que la neuropathologie a rempli historiquement son rôle et qu'il est temps aujourd'hui de passer à d'autres techniques pour élucider la cause de la maladie d'Alzheimer et envisager son traitement. Il faut certes utiliser et perfectionner toutes les méthodes à notre disposition – et la place donnée aujourd'hui à la biologie moléculaire n'est pas usurpée – mais il faut continuer à examiner les cerveaux des patients et des sujets considérés comme intellectuellement normaux même si la législation a édicté des règles strictes concernant l'étude scientifique de prélèvements humains. Celles-ci ont conduit à modifier la procédure permettant d'avoir accès au tissu et des campagnes de « dons du cerveau pour la recherche » ont été initiées. Au moins deux constatations conduisent à cette conclusion : nombre de questions que la neuropathologie de la maladie d'Alzheimer continue de poser demeurent non résolues ; l'expérience d'autres maladies neurodégénératives (comme les ataxies cérébelleuses, les syndromes parkinsoniens ou tout récemment la sclérose latérale amyotrophique et la démence fronto-temporale) montre que l'étude directe du tissu cérébral peut modifier en quelques années la façon dont nous appréhendons la pathologie. Il est très important de déterminer aujourd'hui des valeurs de prévalence des lésions en fonction de l'âge et des populations étudiées afin de pouvoir analyser l'efficacité des mesures de prévention ou des nouvelles thérapeutiques. Ces études en population n'ont pas encore été réalisées en France et l'ont été très peu dans le monde : on peut citer « l'étude des Nonnes » où toutes les sœ urs de Notre-Dame appartenant aux communautés établies aux États-Unis ont accepté de passer des tests cognitifs de façon prospective et ont accepté d'être autopsiées le moment venu (Riley et coll., 2002
), et l'étude MRC-CFAS en Angleterre (
Neuropathology Group et
Medical Research Council Cognitive Function and Aging Study, 2001
). Ces deux études font état d'un grand nombre de lésions vasculaires et mentionnent la fréquence des plaques séniles et des DNF chez des sujets considérés comme intellectuellement normaux, ou peu affectés. Les critères diagnostiques actuels prennent mal en compte la prévalence élevée des lésions dans la population générale et sous-estiment probablement la fréquence des « normaux-malades ».
Lésions associées
Les premières corrélations anatomo-cliniques ont été réalisées chez des patients vivant dans des services de long séjour. Comme nous venons de le signaler, des études plus récentes faites en population ont montré la fréquence des lésions asymptomatiques ou bien tolérées dans la population (
Neuropathology Group et
Medical Research Council Cognitive Function and Aging Study, 2001
; Riley et coll., 2002
). C'est ainsi que dans 19 % des cas « sans démence » était notée la présence de lésions en nombre suffisant pour porter le diagnostic de maladie d'Alzheimer dans l'étude du MRC-CFAS.
Lésions vasculaires
La fréquence des lésions vasculaires (27 % dans le groupe sans démence de la même étude, 16 % dans le groupe avec démence) est plus inattendue. On sait que ces lésions abaissent le seuil à partir duquel les lésions d'Alzheimer sont symptomatiques. Le rapport entre les lésions vasculaires et celles de la maladie d'Alzheimer est discuté : pour certains, le lien est causal (de la Torre, 1994
). S'il est bien établi que le traitement anti-hypertenseur réduit l'incidence de la démence (Forette et coll., 2002
) ou que les facteurs de risque vasculaires sont associés à une prévalence plus élevée des troubles cognitifs, la preuve d'une interaction directe entre les deux pathologies n'a pas encore été apportée. Il a, en revanche, été montré qu'à déficit cognitif égal, la densité des lésions d'Alzheimer était plus basse lorsque des lésions vasculaires y étaient associées, ce qui suggère un abaissement du seuil symptomatique (Zekry et coll., 1997
, 2002
et 2003
).
Maladie de Parkinson et démence à corps de Lewy
Les relations entre la démence d'Alzheimer et la démence à corps de Lewy ou la maladie de Parkinson ont fait couler beaucoup d'encre : initialement considérée comme la cause des troubles cognitifs observés dans la maladie de Parkinson (Boller et coll., 1980
), il ne semble pas, en réalité, que la maladie d'Alzheimer chez les malades atteints de Parkinson soit plus fréquente que dans la population générale (Apaydin et coll., 2002
). Cependant, il semble exister une interaction entre les deux pathologies, puisque la densité des corps de Lewy et celle des plaques séniles ou des dégénérescences neurofibrillaires sont statistiquement liées dans le cortex (Apaydin, et coll., 2002
).
Maladie des grains
La maladie des grains est caractérisée par la présence dans l'hippocampe et l'amygdale de structures ovoïdes de quelques mm de diamètre, mises en évidence par l'imprégnation argentique de Gallyas ou le marquage immunohistochimique de la protéine Tau (Braak et Braak, 1987
). Il s'agit d'accumulation de protéine Tau 4R dans les synapses (Togo et coll., 2002
). La signification de ces lésions, fortement liées à l'âge (Ding et coll., 2006
), reste encore discutée (pour revue voir Probst et Tolnay, 2002
). Elles semblent pouvoir, à elles seules, expliquer certains déficits cognitifs mais elles sont le plus souvent associées aux lésions de la maladie d'Alzheimer.
Rapidité de l'évolution
Comme nous l'avons mentionné, les plaques séniles et les DNF sont rares chez les sujets jeunes ; elles sont constantes chez les centenaires. Deux opinions ont été exprimées à ce sujet : pour certains, les DNF sont « normales » à partir d'un certain âge ; elles sont le témoin du « vieillissement physiologique ». Pour Braak et Braak, au contraire, elles sont toujours le témoin d'une maladie d'Alzheimer dont l'évolution est inéluctable. Il est impossible aujourd'hui de déterminer la rapidité de constitution des lésions. Il est douteux que la moitié de la population porteuse de DNF à 47 ans souffre un jour d'une maladie d'Alzheimer clinique. Les méthodes neuropathologiques ne permettent pas d'avoir une vision dynamique des lésions : progressent-elles toutes au même pas ? Au contraire, peuvent-elles rester stables pendant des années, des décennies ? Régressent-elles parfois ? Il faudra sans doute attendre la visualisation des lésions in vivo ou l'apparition de marqueurs biologiques sensibles et spécifiques pour mieux comprendre l'évolution ou les évolutions lésionnelle(s).
Relation entre lésions liées à l'accumulation de peptide Aβet celles dues à l'accumulation de Tau
Dans l'hypothèse de la cascade amyloïde, les accumulations intracellulaires de protéine Tau sont la conséquence de l'augmentation de production de peptide Aβ. Si le rôle essentiel du peptide Aβdans la physiopathologie est généralement accepté, plusieurs éléments de la cascade amyloïde restent discutés.
Précession des lésions de DNF
Comme nous l'avons vu, dans la grande série de cas neuropathologiques étudiés par Braak et Braak (1997
), une proportion significative de cas présentait des DNF en l'absence de dépôts de peptide Aβ, ce qui a été interprété comme la preuve que les premières apparaissaient avant les secondes, mais il est vrai qu'il est difficile de démontrer qu'aucun dépôt de peptide Aβn'est présent dans le cerveau. Il faudrait multiplier les prélèvements et échantillonner le cortex de façon systématique et extensive pour réduire au maximum le nombre d'aires corticales non analysées, une étude qui reste à faire.
Impossibilité de reproduire la DNF par simple surexpression de peptide Aβchez les animaux modèles
Aucune DNF n'est constatée, même à un âge avancé, chez les souris transgéniques qui surexpriment l'APP humain muté, parfois en association avec le gène muté de la préséniline 1. Tout au plus peut-on observer la présence de protéine Tau hyperphosphorylée (sans agrégation) autour des plaques séniles. Dans ces modèles, la dégénérescence neurofibrillaire n'apparaît pas comme la conséquence directe de l'accumulation de peptide Aβ.
Les souris transgéniques Tau développent des DNF. Elles ont été croisées avec des souris APP. Des accumulations intracellulaires de Tau et extracellulaires de peptide Aβont ainsi pu être obtenues, mais l'interaction entre les deux types de lésions était limitée à une augmentation du nombre des DNF dans les systèmes olfactifs et limbique chez la souris double transgénique (Lewis et coll., 2001
). Les gènes mutés de l'APP, de la préséniline 1 et de Tau sont présents dans la souris triple transgénique développée par le groupe de LaFerla (Oddo et coll., 2003
). Des plaques séniles et des DNF sont observées ; l'apparition des dépôts de peptide Aβprécède celui des DNF. L'interaction entre l'accumulation de Tau et de peptide Aβet son mécanisme restent à élucider. L'injection de peptide Aβdans un modèle transgénique Tau a conduit à l'apparition préférentielle ou précoce de DNF dans les neurones qui projetaient sur la région où le peptide Aβavait été injecté (Götz et coll., 2001
). Il y a donc des indices d'une interaction entre les dépôts amyloïdes et l'accumulation et l'agrégation de la protéine Tau, mais les mécanismes impliqués dans cette interaction sont encore peu connus.
En conclusion,
la neuropathologie de la maladie d'Alzheimer est loin d'avoir livré tous ses secrets. Ses progrès ont été, ces 20 dernières années, spectaculaires : l'identification du peptide Aβet de la protéine Tau au sein des lésions, par exemple, est à mettre en partie à son actif. Aujourd'hui, l'examen de tissu humain post mortem n'est plus la seule ressource dont le neuropathologiste dispose. L'analyse de modèles dynamiques, comme ceux obtenus par transgenèse chez la souris, a déjà modifié notre compréhension. Demain, la visualisation des lésions in vivo nous permettra de mieux appréhender l'évolution de la maladie. Dans un avenir proche, le rôle des oligomères de peptide Aβ, les interactions entre le peptide Aβet la protéine Tau, le contenu lipidique des plaques séniles, les effets des traitements sont autant de sujets de recherche que la neuropathologie contribuera à éclairer, avec un corollaire : il faut continuer à pratiquer des autopsies de patients et de sujets témoins, et favoriser les programmes de « don de cerveau pour la recherche scientifique », en particulier en population.
Bibliographie
[1] abraham cr,
selkoe dj,
potter h. Immunochemical identification of the serine protease inhibitor a-1 antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease.
Cell. 1988;
52:487
-501

[2] achim c,
auer r,
bergeron c,
cardozo a,
deprez m. Global democratic consensus on neuropathological disease criteria.
Lancet Neurol. 2002;
1:340
[3] alzheimer a. Uber eine eigenartige Erkrankung der Hirnrinde.
Allgemeine Zeitschr Psychiatr Gerichtlisch Med. 1907;
64:146
-148
[4] anderton bh,
breinburg d,
downes mj,
green pj,
tomlinson be. Monoclonal antibodies show that neurofibrillary tangles and neurofilaments share antigenic determinants.
Nature. 1982;
298:84
-86
[5] apaydin h,
ahlskog je,
parisi je,
boeve bf,
dickson dw. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response.
Arch Neurol. 2002;
59:102
-112
[6] arends ym,
duyckaerts c,
rozemuller jm,
eikelenboom p,
hauw jj. Microglia, amyloid and dementia in Alzheimer disease. A correlative study.
Neurobiol Aging. 2000;
21:39
-47
[7] attems j,
jellinger ka. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology--a pilot study.
Acta Neuropathol (Berl). 2004;
107:83
-90
[8] ball m,
braak h,
coleman p,
dickson d,
duyckaerts c. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease.
Neurobiology of Aging. 1997;
18:S1
-S2
[9] bancher c,
brunner c,
lassmann h,
budka h,
jellinger k. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease.
Brain Res. 1989;
477:90
-99
[10] berg l,
mckeel dwj,
miller jp,
storandt m,
rubin eh. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype.
Arch Neurol. 1998;
55:326
-335
[11] berger b,
tassin jp,
rancurel g,
blanc g. Catecholaminergic innervation of the human cerebral cortex in presenile and senile dementia : histochemical and biochemical studies.
In: usdin e, sourkeset tl, youdim mbh, editors.
Enzymes and neurotransmitters in mental disease..
Chichester:John Wiley & Sons;
1980.
p. 317
-328
[12] bielschowsky m. Die Silberimprägnation der Achsencylinder.
Neurologisches Zentralblatt (Leipzig). 1902;
21:579
-584
[13] bielschowsky m. Der Silberimprägnation der Neurofibrillen.
Neurol Centralbl. 1903;
22:997
-1006
[14] boller f,
mizutani r,
roessmann u,
gambetti p. Parkinson’s disease, dementia and Alzheimer’s disease: clinico-pathological correlations.
Ann Neurol. 1980;
1:329
-355
[15] boncristiano s,
calhoun me,
howard v,
bondolfi l,
kaeser sa. Neocortical synaptic bouton number is maintained despite robust amyloid deposition in APP23 transgenic mice.
Neurobiol Aging. 2005;
26:607
-613
[16] braak h,
braak e. Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes.
Neurosc Lett. 1987;
76:124
-127
[17] braak h,
braak e. Neuropil threads occur in dendrites of tangle-bearing nerve cells.
Neuropathol Appl Neurobiol. 1988;
14:39
-44
[18] braak h,
braak e. Neuropathological stageing of Alzheimer-related changes.
Acta Neuropathol (Berl). 1991;
82:239
-259
[19] braak h,
braak e. Frequency of stages of Alzheimer-related lesions in different age categories.
Neurobiol Aging. 1997;
18:351
-357
[20] braak h,
braak e,
grundke-iqbal i,
iqbal k. Occurence of neuropil threads in the senile human brain and in Alzheimer’s disease. A 3rd location of paired helical filaments outside of neurofilament tangles and neuritic plaques.
Neurosci Lett. 1986;
65:351
-355
[21] brilliant mj,
elble rj,
ghobrial m,
struble rg. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease.
Neuropathol Appl Neurobiol. 1997;
23:322
-325
[22] brion jp,
passareiro h,
nunez j,
flament-durand j. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer.
Arch Biol (Brux). 1985;
95:229
-235
[23] buldyrev sv,
cruz l,
gomez-isla t,
gomez-tortosa e,
havlin s. Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias.
Proc Natl Acad Sci USA. 2000;
97:5039
-5043
[24] cleveland dw,
hwo sy,
kirschner mw. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin.
J Mol Biol. 1977;
116:207
-225
[25] cotman cw. Apoptosis decision cascades and neuronal degeneration in Alzheimer’s disease.
Neurobiol Aging. 1998;
19:S29
-S32
[26] cotman cw,
anderson aj. A potential role for apoptosis in neurodegeneration and Alzheimer’s disease.
Mol Neurobiol. 1995;
10:19
-45
[27] cras p,
smith ma,
richey pl,
siedlak sl,
mulvihill p,
perry g. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease.
Acta Neuropathol (Berl). 1995;
89:291
-295
[28] crook r,
verkkoniemi a,
perez-tur j,
mehta n,
baker m. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1.
Nat Med. 1998;
4:452
-455
[29] de la torre jc. Impaired brain microcirculation may trigger Alzheimer’s disease.
Neurosci Biobehav. Rev 1994;
18:397
-401
[30] decarli c,
kaye ja,
horwitz b,
rapoport si. Critical analysis of the use of computer-assisted transverse axial tomography to study human brain in aging and dementia of the Alzheimer type.
Neurology. 1990;
40:872
-883
[31] delacourte a,
david jp,
sergeant n,
buee l,
wattez a. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease.
Neurology. 1999;
52:1158
-1165
[32] delaère p,
duyckaerts c,
masters c,
piette f,
hauw jj. Large amounts of neocortical βA4 deposits without Alzheimer changes in a nondemented case.
Neurosci Lett. 1990;
116:87
-93
[33] delaère p,
duyckaerts c,
he y,
piette f,
hauw jj. Subtypes and differential laminar distributions of βA4 deposits in Alzheimer’s disease : relationship with the intellectual status of 26 cases.
Acta Neuropathol (Berl). 1991;
81:328
-335
[34] delaère p,
he y,
fayet g,
duyckaerts c,
hauw jj. Beta A4 deposits are constant in the brain of the oldest old: an immunocytochemical study of 20 French centenarians.
Neurobiol Aging. 1993;
14:191
-194
[35] delatour b,
blanchard v,
pradier l,
duyckaerts c. The innervation of senile plaques: a link between amyloid and neurofibrillary pathology?.
In: gauthier s, scheltens p, cummings jl, editors.
Alzheimer’s disease and related disorders. Annual 2004..
London:Martin Dunitz;
2003.
p. 1
-19
[36] dickson dw,
crystal ha,
mattiace la,
masur dm,
blau ad. Identification of normal and pathological aging in prospectively studied nondemented elderly humans.
Neurobiol Aging. 1991;
13:179
-189
[37] dickson dw,
lee sc,
mattiace la,
yen sc,
brosnan c. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease.
Glia. 1993;
7:75
-83
[38] dickson dw,
crystal ha,
bevona c,
honer w,
vincent i,
davies p. Correlations of synaptic and pathological markers with cognition of the elderly.
Neurobiol Aging. 1995;
16:285
-304
[39] dickson tc,
saunders hl,
vickers jc. Relationship between apolipoprotein E and the amyloid deposits and dystrophic neurites of Alzheimer’s disease.
Neuropathol Appl Neurobiol. 1997;
23:483
-491
[40] ding zt,
wang y,
jiang yp,
yoshida m,
mimuro m,
inagaki t. Argyrophilic grain disease: frequency and neuropathology in centenarians.
Acta Neuropathol (Berl). 2006;
111:320
-328
[41] divry p. Etude histochimique des plaques séniles.
J Belge Neurol Psych. 1927;
9:643
-657
[42] duyckaerts c,
dickson d. Neuropathology of Alzheimer’s disease.
In: dickson d, editors.
Neurode-generationnbsp;:nbsp;the molecular pathology of dementia and mouvement disorders.
Basel:ISN Neuropath Press;
2003.
p. 47
-65
[43] duyckaerts c,
hauw jj. Diagnosis and staging of Alzheimer disease.
Neurobiol Aging. 1997a;
18:S33
-42
[44] duyckaerts c,
hauw jj. Prevalence, incidence and duration of Braak’s stages in the general population: can we know?.
Neurobiol Aging. 1997b;
18:362
-369, discussion 389-392
[45] duyckaerts c,
hauw jj,
piette f,
rainsard c,
poulain v. Cortical atrophy in senile dementia of the Alzheimer type is mainly due to a decrease in cortical length.
Acta Neuropathol (Berl). 1985;
66:72
-74
[46] duyckaerts c,
hauw jj,
bastenaire f,
piette f,
poulain c. Laminar distribution of neocortical plaques in senile dementia of the Alzheimer type.
Acta Neuropathol (Berl). 1986;
70:249
-256
[47] duyckaerts c,
kawasaki h,
delaère p,
rainsard c,
hauw jj. Fiber disorganization in the neocortex of patients with senile dementia of the Alzheimer type.
Neuropath Appl Neurobiol. 1989a;
15:233
-247
[48] duyckaerts c,
llamas e,
delaère p,
miele p,
hauw jj. Neuronal loss and neuronal atrophy. Computer simulation in connection with Alzheimer’s disease.
Brain Res. 1989b;
504:94
-100
[49] duyckaerts c,
delaère p,
he y,
camilleri s,
braak h. The relative merits of tau- and amyloid markers in the neuropathology of Alzheimer’s disease.
In: bergener m, finkel si, editors.
Treating Alzheimer’s and other dementias.
New York:Springer;
1995.
p. 81
-89
[50] duyckaerts c,
bennecib m,
grignon y,
uchihara t,
he y. Modeling the relation between neurofibrillary tangles and intellectual status.
Neurobiol Aging. 1997;
18:267
-273
[51] forette f,
seux ml,
staessen ja,
thijs l,
babarskiene mr. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study.
Arch Intern Med. 2002;
162:2046
-2052
[52] frackowiak j,
wisniewski hm,
wegiel j,
merz gs,
iqbal k,
wang kc. Ultrastructure of the microglia that phagocytes amyloid and the microglia that produces beta-amyloid fibrils.
Acta Neuropathol (Berl). 1992;
84:225
-223
[53] gearing m,
levey ai,
mirra ss. Diffuse plaques in the striatum in Alzheimer disease (AD): relationship to the striatal mosaic and selected neuropeptide markers.
J Neuropathol Exp Neurol. 1997;
56:1363
-1370
[54] giannakopoulos p,
gold g,
kovari e,
von gunten a,
imhof a. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience.
Acta Neuropathol (Berl). 2007;
113:1
-12
[55] glenner gg. Amyloid deposits and amyloidosis. The β-fibrilloses.
N Engl J Med. 1980;
302:1283
-1292
[56] glenner gg,
wong cw. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein.
Biochem Biophys Res Commun. 1984;
120:885
-890
[57] goedert m,
wischik cm,
crowther ra,
walker je,
klug a. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau.
Proc Natl Acad Sci USA. 1988;
85:4051
-4055
[58] goedert m,
spillantini mg,
potier mc,
ulrich j,
crowther ra. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain.
Embo J. 1989;
8:393
-399
[59] gomez-isla t,
price jl,
mckeel dw,
morris jc,
growdon jh,
hyman bt. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease.
J Neurosci. 1996;
16:4491
-4500
[60] gomez-isla t,
hollister r,
west h,
mui s,
growdon jh,
petersen rc. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease.
Ann Neurol. 1997;
41:17
-24
[61] götz j,
chen f,
van dorpe j,
nitsch rm. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Abeta42 fibrils.
Science. 2001;
293:1491
-1495
[62] gouras gk,
tsai j,
naslund j,
vincent b,
edgar m. Intraneuronal Abeta42 accumulation in human brain.
Am J Pathol. 2000;
156:15
-20
[63] grignon y,
duyckaerts c,
bennecib m,
hauw jj. Cytoarchitectonic alterations in the supramarginal gyrus of late onset Alzheimer’s disease.
Acta Neuropathol (Berl). 1998;
95:395
-406
[64] grundke-iqbal i,
iqbal k,
tung yc,
quinlan m,
wisniewski hm,
binder li. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology.
Proc Natl Acad Sci USA. 1986;
83:4913
-4917
[65] hansen la,
deteresa r,
davies p,
terry rd. Neocortical morphometry, lesion counts, and choline acetyltransferase levels in the age spectrum of Alzheimer’s disease.
Neurology. 1988;
38:48
-54
[66] hardy j. An ’anatomical cascade hypothesis’ for Alzheimer’s disease.
Trends in Neurosciences. 1992;
15:200
-201
[67] hauw jj,
vignolo p,
duyckaerts c,
beck m,
forette f. Etude neuropathologique de 12 centenaires: la fréquence de la démence sénile de type Alzheimer n’est pas particulièrement élevée dans ce groupe de personnes très âgées.
Rev Neurol (Paris). 1986;
142:107
-115
[68] he y,
delaère p,
duyckaerts c,
wasowicz m,
piette f,
hauw jj. Two distinct ubiquitin immunoreactive senile plaques in Alzheimer’s disease: relationship with the intellectual status in 29 cases.
Acta Neuropathol. 1993;
86:109
-116
[69] hubbard bm,
anderson jm. Age, senile dementia and ventricular enlargement.
J Neurol Neurosurg Psychiat. 1981;
44:631
-635
[70] iseki e,
matsushita m,
kosaka k,
kondo h,
ishii t,
amano n. Distribution and morphology of brain stem plaques in Alzheimer’s disease.
Acta Neuropathol (Berl). 1989;
78:131
-136
[71] joachim cl,
morris jh,
selkoe dj. Amyloid angiopathy in 100 cases of Alzheimer’s disease.
Neurology. 1987;
371:225
[72] joachim cl,
morris jh,
selkoe dj. Clinically diagnosed Alzheimer’s disease: autopsy results in 150 cases.
Ann Neurol. 1988;
24:50
-56
[73] joachim cl,
morris jh,
selkoe dj. Diffuse amyloid plaques occur commonly in the cerebellum in Alzheimer’s disease.
Am J Pathol. 1989;
135:309
-319
[74] kang j,
lemaire h-g,
unterbeck a,
salbaum jm,
masters cl. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor.
Nature. 1987;
325:733
-736
[75] khachaturian zs. Diagnosis of Alzheimer’s disease.
Archives of Neurology. 1985;
42:1097
-1105
[76] knobloch m,
konietzko u,
krebs dc,
nitsch rm. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice.
Neurobiol Aging. 2006 Jul 28;
[Epub ahead of print]:
-
[77] knowles rb,
wyart c,
buldyrev sv,
cruz l,
urbanc b. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease.
Proc Natl Acad Sci USA. 1999;
96:5274
-5279
[78] kumar-singh s,
cras p,
wang r,
kros jm,
van swieten j. Dense-core senile plaques in the Flemish variant of Alzheimer’s disease are vasocentric.
Am J Pathol. 2002;
161:507
-520
[79] lacor pn,
buniel mc,
furlow pw,
clemente as,
velasco pt. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease.
J Neurosci. 2007;
27:796
-807
[80] langui d,
girardot n,
el hachimi h,
allinquant b,
blanchard v. Subcellular topography of neuronal A-beta peptide in APPxPS1 transgenic mice.
Am J Pathol. 2004;
165:1465
-1477
[81] le tv,
crook r,
hardy j,
dickson dw. Cotton wool plaques in non-familial late-onset Alzheimer disease.
J Neuropathol Exp Neurol. 2001;
60:1051
-1061
[82] levy e,
carman md,
fernandez-madrid ij,
power md,
lieberburg i. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type.
Science. 1990;
248:1124
-1126
[83] lewis j,
dickson dw,
lin wl,
chisholm l,
corral a. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP.
Science. 2001;
293:1487
-1491
[84] liu k,
solano i,
mann d,
lemere c,
mercken m. Characterization of Abeta11-40/42 peptide deposition in Alzheimer’s disease and young Down’s syndrome brains: implication of N-terminally truncated Abeta species in the pathogenesis of Alzheimer’s disease.
Acta Neuropathol (Berl). 2006;
112:163
-74
[85] mann dma,
esiri mm. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome.
J Neurol Sci. 1989;
89:169
-179
[86] mann dm,
jones d,
prinja d,
purkiss ms. The prevalence of amyloid (A4) protein deposits within the cerebral and cerebellar cortex in Down’s syndrome and Alzheimer’s disease.
Acta Neuropathol (Berl). 1990;
80:318
-327
[87] masliah e,
terry rd. Role of synaptic pathology in the mechanisms of denervation in Alzheimer disease.
Clin Neurosci. 1993;
4:192
-198
[88] masliah e,
terry rd,
deteresa rm,
hansen la. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease.
Neurosci Lett. 1989;
103:234
-239
[89] masliah e,
mallory m,
hansen l,
deteresa r,
terry rd. Quantitative synaptic alterations in the human neocortex during normal aging.
Neurology. 1993;
43:192
-197
[90] masliah e,
mallory m,
hansen l,
deteresa r,
alford m,
terry r. Synaptic and neuritic alterations during the progression of Alzheimer’s disease.
Neurosci Lett. 1994;
174:67
-72
[91] mattson m,
keller j,
begley j. Evidence for synaptic apoptosis.
Exp Neurol. 1998;
153:35
-48
[92] mcgeer pl,
akiyama h,
itagaki s,
mcgeer eg. Complement activation in amyloid plaques in Alzheimer’s dementia.
Neurosci Letters. 1989;
107:341
-346
[93] mckee ac,
au r,
cabral hj,
kowall nw,
seshadri s. Visual association pathology in preclinical Alzheimer disease.
J Neuropathol Exp Neurol. 2006;
65:621
-630
[94] mckhann g,
drachman d,
folstein m,
katzman r,
price d,
stadlan em. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease.
Neurology. 1984;
19:939
-944
[95] metsaars wp,
hauw jj,
welsem me,
duyckaerts c. A grading system of Alzheimer disease lesions in neocortical areas.
Neurobiol Aging. 2003;
24:563
-572
[96] mirra ss,
heyman a,
mckeel d,
sumi sm,
crain bj. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathological assessment of Alzheimer’s disease.
Neurology. 1991;
41:479
-486
[97] morel f. Petite contribution à l’étude d’une angiopathie apparemment dyshorique et topistique.
Mschr Psychiat Neurol. 1950;
120:352
-357
[98] mori t,
paris d,
town t,
rojiani am,
sparks dl. Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice.
J Neuropathol Exp Neurol. 2001;
60:778
-785
[99] nagy z,
esiri mm,
smith ad. The cell division cycle and the pathophysiology of Alzheimer’s disease.
Neuroscience. 1998;
87:731
-739
[100] najlerahim a,
bowen dm. Regional weight loss in the cerebral cortex and some suncortical nuclei in senile dementia of the Alzheimer type.
Acta Neuropathol (Berl). 1989;
75:509
-512
[101]neuropathology group, medical research council cognitive function and aging study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS).
Lancet. 2001;
357:169
-175
[102] oddo s,
caccamo a,
shepherd jd,
murphy mp,
golde te. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction.
Neuron. 2003;
39:409
-421
[103] probst a,
tolnay m. Argyrophilic grain disease (AgD), a frequent and largely underestimated cause of dementia in old patients.
Rev Neurol (Paris). 2002;
158:155
-165
[104] regeur l,
badsberg jensen g,
pakkenberg h,
evans sm,
pakkenberg b. No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer’s type.
Neurobiol Aging. 1994;
15:347
-352
[105] riley kp,
snowdon da,
markesbery wr. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study.
Ann Neurol. 2002;
51:567
-577
[106] rohn tt,
head e,
su jh,
anderson aj,
bahr ba. Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer’s disease.
Am J Pathol. 2001;
158:189
-198
[107] rozemuller aj,
roos ra,
bots gt,
kamphorst w,
eikelenboom p,
van nostrand we. Distribution of beta/A4 protein and amyloid precursor protein in hereditary cerebral hemorrhage with amyloidosis-Dutch type and Alzheimer’s disease.
Am J Pathol. 1993;
142:1449
-1457
[108] rub u,
del tredici k,
del turco d,
braak h. The intralaminar nuclei assigned to the medial pain system and other components of this system are early and progressively affected by the Alzheimer’s disease-related cytoskeletal pathology.
J Chem Neuroanat. 2002;
23:279
-290
[109] russo c,
salis s,
dolcini v,
venezia v,
song xh. Amino-terminal modification and tyrosine phosphorylation of [corrected] carboxy-terminal fragments of the amyloid precursor protein in Alzheimer’s disease and Down’s syndrome brain.
Neurobiol Dis. 2001;
8:173
-180
[110] sassin i,
schultz c,
thal dr,
rub u,
arai k. Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert.
Acta Neuropathol (Berl). 2000;
100:259
-269
[111] scheff sw,
price da. Synapse loss in the temporal lobe in Alzheimer’s disease.
Ann Neurol. 1993;
33:190
-199
[112] scheff sw,
price da. Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus.
J Alzheimers Dis. 2006;
9:101
-115
[113] scheff sw,
dekosky st,
price da. Quantitative assessment of cortical synaptic density in Alzheimer’s disease.
Neurobiol Aging. 1990;
11:29
-37
[114] scheff sw,
sparks l,
price da. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease.
Ann Neurol. 1993;
34:356
-361
[115] scheff sw,
sparks dl,
price da. Quantitative assessment of synaptic density in the outer molecular layer of the hippocampal dentate gyrus in Alzheimer’s disease.
Dementia. 1996;
7:226
-232
[116] schenk d,
barbour r,
dunn w,
gordon g,
grajeda h. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse.
Nature. 1999;
400:173
-177
[117] schmidt m,
lee v,
trojanowski j. Comparative epitope analysis of neuronal cytoskeletal proteins in Alzheimer’s disease senile plaque, neurites and neuropil threads.
Lab Invest. 1991;
64:352
-357
[118] seubert p,
vigo-pelfrey c,
esch f,
lee m,
dovey h. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids.
Nature. 1992;
359:325
-327
[119] shimohama s,
kamiya s,
taniguchi t,
akagawa k,
kimura j. Differential involvement of synaptic vesicle and presynaptic plasma membrane proteins in Alzheimer’s disease.
Biochem Biophys. Res Commun 1997;
236:239
-242
[120] soderberg l,
bogdanovic n,
axelsson b,
winblad b,
naslund j,
tjernberg lo. Analysis of single Alzheimer solid plaque cores by laser capture microscopy and nanoelectrospray/tandem mass spectrometry.
Biochemistry. 2006;
45:9849
-9856
[121] stadelmann c,
bruck w,
bancher c,
jellinger k,
lassmann h. Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis.
J Neuropathol Exp Neurol. 1998;
57:456
-464
[122] suzuki n,
iwatsubo t,
odaka a,
ishibashi y,
kitada c,
ihara y. High tissue content of soluble β1-40 is linked to cerebral amyloid angiopathy.
Am J Pathol. 1994;
145:452
-460
[123] tagliavini f,
giaccone g,
frangione b,
bugiani p. Preamyloid deposits in the cerebral cortex of patients with Alzheimer’s disease and nondemented individuals.
Neurosci Lett. 1988;
93:191
-196
[124] tekirian tl,
saido tc,
markesbery wr,
russell mj,
wekstein dr. N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits.
J Neuropathol Exp Neurol. 1998;
57:76
-94
[125] terry rd,
peck a,
deteresa r,
schechter r. Some morphometric aspects of the brain in senile dementia of the Alzheimer type.
Ann Neurol. 1981;
10:184
-192
[126] terry rd,
masliah e,
salmon dp,
butters n,
deteresa r. Physical basis of cognitive alterations in Alzheimer’s disease : synapse loss is the major correlate of cognitive impairment.
Ann Neurol. 1991;
30:572
-580
[127] terry d,
masliah e,
hansen la. Structural basis of the cognitive alterations in Alzheimer disease.
In: terry rd, katzman r, bick kl, editors.
Alzheimer disease..
New York:Raven Press;
1994.
p. 179
-196
[128] thal dr,
sassin i,
schultz c,
haass c,
braak e,
braak h. Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Abeta.
Neuropathol Exp Neurol. 1999;
58:210
-216
[129] thal dr,
rub u,
orantes m,
braak h. Phases of A beta-deposition in the human brain and its relevance for the development of AD.
Neurology. 2002;
58:1791
-1800
[130] togo t,
sahara n,
yen sh,
cookson n,
ishizawa t. Argyrophilic grain disease is a sporadic 4-repeat tauopathy.
J Neuropathol Exp Neurol. 2002;
61:547
-556
[131] troncoso jc,
cataldo am,
nixon ra,
barnett jl,
lee mk. Neuropathology of preclinical and clinical late-onset Alzheimer’s disease.
Ann Neurol. 1998;
43:673
-676
[132] uchihara t,
duyckaerts c,
he y,
kobayashi k,
seilhean d. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain.
Neurosci Lett. 1995;
195:5
-8
[133] vidal r,
calero m,
piccardo p,
farlow mr,
unverzagt fw. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele.
Acta Neuropathol (Berl). 2000;
100:1
-12
[134] walsh dm,
klyubin i,
fadeeva jv,
cullen wk,
anwyl r. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo.
Nature. 2002;
416:535
-539
[135] wang q,
walsh dm,
rowan mj,
selkoe dj,
anwyl r. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5.
J Neurosci. 2004;
24:3370
-3378
[136] wegiel j,
kuchna i,
nowicki k,
frackowiak j,
mazur-kolecka b. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration.
Acta Neuropathol (Berl). 2007;
113:389
-402
[137] wilcock gk,
esiri mm. Plaques, tangles and dementia. A quantitative study.
J Neurol Sci. 1982;
56:343
-356
[138] wisniewski hm,
sadowski m,
jakubowska-sadowska k,
tarnawski m,
wegiel j. Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease.
J Neuropathol Exp Neurol. 1998;
57:674
-683
[139] zekry d,
duyckaerts c,
belmin j,
geoffre c,
vlaicu m. Vascular, degenerative and mixed dementia. A clinicopathological study in 33 cases.
Brain Pathol. 1997;
7:1213
[140] zekry d,
duyckaerts c,
moulias r,
belmin j,
geoffre c. Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly.
Acta Neuropathol (Berl). 2002;
103:481
-487
[141] zekry d,
duyckaerts c,
belmin j,
geoffre c,
herrmann f. The vascular lesions in vascular and mixed dementia: the weight of functional neuroanatomy.
Neurobiol Aging. 2003;
24:213
-219
→ Aller vers SYNTHESE