Connaissances fondamentales
2007
| ANALYSE |
2-
Cascade amyloïde
Ce n'est que récemment que la biologie moléculaire a permis d'obtenir des avancées significatives dans la compréhension des mécanismes sous-tendant les processus de formation des lésions de la maladie d'Alzheimer. Ainsi, la description de la nature du composant majeur des plaques séniles, ces agrégats protéiques qui s'accumulent au cours de la maladie, ne date que de 1984 avec l'avènement du maintenant célèbre peptide amyloïde (peptide Aβ) (Glenner et Wong, 1984a et b ; Masters et coll., 1985). C'est trois ans plus tard que le précurseur de ce peptide, la βAPP (β-Amyloid Precursor Protein), a été cloné (Goldgaber et coll., 1987 ; Kang et coll., 1987 ; Tanzi et coll., 1987). Par la suite, il est apparu que certaines formes agressives et à début précoce de maladie d'Alzheimer étaient d'origine génétique et on a identifié les loci des gènes impliqués sur les chromosomes 21, 14 et 1 (pour revues voir : Tanzi et coll., 1991 ; Mullan et Crawford, 1993 ; Schellenberg, 1995). La biologie moléculaire a permis d'identifier les protéines responsables de ces formes précoces ; la βAPP et les présénilines 1 et 2 (Levy-Lahad et coll., 1995 ; Rogaev et coll., 1995 ; Sherrington et coll., 1995 ; Alzheimer's Disease Collaborative Group, 1996). Ces découvertes récentes, combinées à l'analyse des phénotypes cellulaires associés aux mutations, ont permis des avancées majeures. Ainsi, il a été établi que l'expression de la βAPP ou des présénilines mutées dans des cellules en culture conduit toujours à une modulation de la production du peptide Aβ. Le fait que des mutations distinctes, portant sur les gènes de protéines différentes et responsables de formes agressives de maladie d'Alzheimer, aient pour dénominateur commun une modulation de la production du peptide Aβ est un argument majeur en faveur de l'hypothèse de la cascade amyloïde qui prédit que l'accumulation de fibrilles amyloïdes conduit, selon une séquence d'événements encore discutée, à la démence caractérisant le tableau clinique terminal des malades (figure 2.1). On peut donc considérer que, même si la surproduction de peptide amyloïde n'est pas stricto sensu le premier déterminant étiologique de la maladie, elle y contribue en tous cas de manière certaine. Ce bilan des connaissances concernera les données récentes sur la maturation physiopathologique de la βAPP et détaillera les avancées concernant les mécanismes de production et de dégradation du peptide Aβ.
et b ; Masters et coll., 1985). C'est trois ans plus tard que le précurseur de ce peptide, la βAPP (β-Amyloid Precursor Protein), a été cloné (Goldgaber et coll., 1987 ; Kang et coll., 1987 ; Tanzi et coll., 1987). Par la suite, il est apparu que certaines formes agressives et à début précoce de maladie d'Alzheimer étaient d'origine génétique et on a identifié les loci des gènes impliqués sur les chromosomes 21, 14 et 1 (pour revues voir : Tanzi et coll., 1991 ; Mullan et Crawford, 1993 ; Schellenberg, 1995). La biologie moléculaire a permis d'identifier les protéines responsables de ces formes précoces ; la βAPP et les présénilines 1 et 2 (Levy-Lahad et coll., 1995 ; Rogaev et coll., 1995 ; Sherrington et coll., 1995 ; Alzheimer's Disease Collaborative Group, 1996). Ces découvertes récentes, combinées à l'analyse des phénotypes cellulaires associés aux mutations, ont permis des avancées majeures. Ainsi, il a été établi que l'expression de la βAPP ou des présénilines mutées dans des cellules en culture conduit toujours à une modulation de la production du peptide Aβ. Le fait que des mutations distinctes, portant sur les gènes de protéines différentes et responsables de formes agressives de maladie d'Alzheimer, aient pour dénominateur commun une modulation de la production du peptide Aβ est un argument majeur en faveur de l'hypothèse de la cascade amyloïde qui prédit que l'accumulation de fibrilles amyloïdes conduit, selon une séquence d'événements encore discutée, à la démence caractérisant le tableau clinique terminal des malades (figure 2.1). On peut donc considérer que, même si la surproduction de peptide amyloïde n'est pas stricto sensu le premier déterminant étiologique de la maladie, elle y contribue en tous cas de manière certaine. Ce bilan des connaissances concernera les données récentes sur la maturation physiopathologique de la βAPP et détaillera les avancées concernant les mécanismes de production et de dégradation du peptide Aβ.
et b ; Masters et coll., 1985). C'est trois ans plus tard que le précurseur de ce peptide, la βAPP (β-Amyloid Precursor Protein), a été cloné (Goldgaber et coll., 1987 ; Kang et coll., 1987 ; Tanzi et coll., 1987). Par la suite, il est apparu que certaines formes agressives et à début précoce de maladie d'Alzheimer étaient d'origine génétique et on a identifié les loci des gènes impliqués sur les chromosomes 21, 14 et 1 (pour revues voir : Tanzi et coll., 1991 ; Mullan et Crawford, 1993 ; Schellenberg, 1995). La biologie moléculaire a permis d'identifier les protéines responsables de ces formes précoces ; la βAPP et les présénilines 1 et 2 (Levy-Lahad et coll., 1995 ; Rogaev et coll., 1995 ; Sherrington et coll., 1995 ; Alzheimer's Disease Collaborative Group, 1996). Ces découvertes récentes, combinées à l'analyse des phénotypes cellulaires associés aux mutations, ont permis des avancées majeures. Ainsi, il a été établi que l'expression de la βAPP ou des présénilines mutées dans des cellules en culture conduit toujours à une modulation de la production du peptide Aβ. Le fait que des mutations distinctes, portant sur les gènes de protéines différentes et responsables de formes agressives de maladie d'Alzheimer, aient pour dénominateur commun une modulation de la production du peptide Aβ est un argument majeur en faveur de l'hypothèse de la cascade amyloïde qui prédit que l'accumulation de fibrilles amyloïdes conduit, selon une séquence d'événements encore discutée, à la démence caractérisant le tableau clinique terminal des malades (figure 2.1). On peut donc considérer que, même si la surproduction de peptide amyloïde n'est pas stricto sensu le premier déterminant étiologique de la maladie, elle y contribue en tous cas de manière certaine. Ce bilan des connaissances concernera les données récentes sur la maturation physiopathologique de la βAPP et détaillera les avancées concernant les mécanismes de production et de dégradation du peptide Aβ.
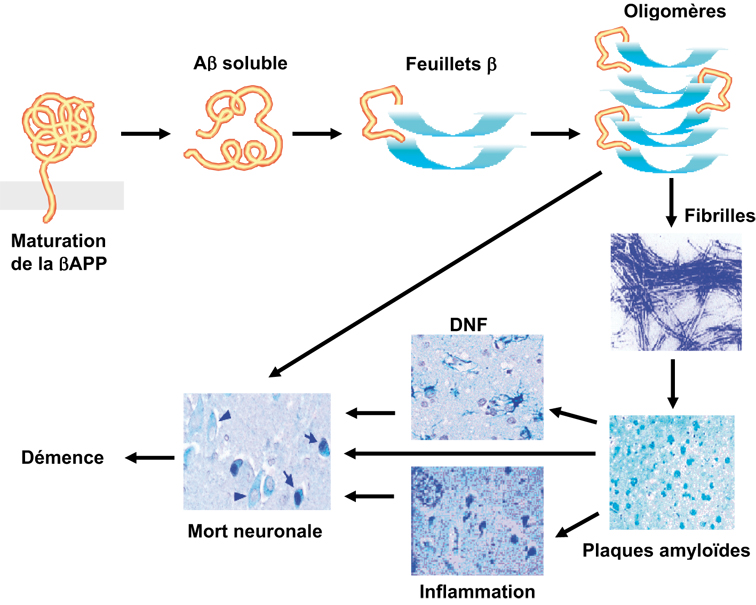
Maturation physiopathologique du précurseur du peptide amyloïde
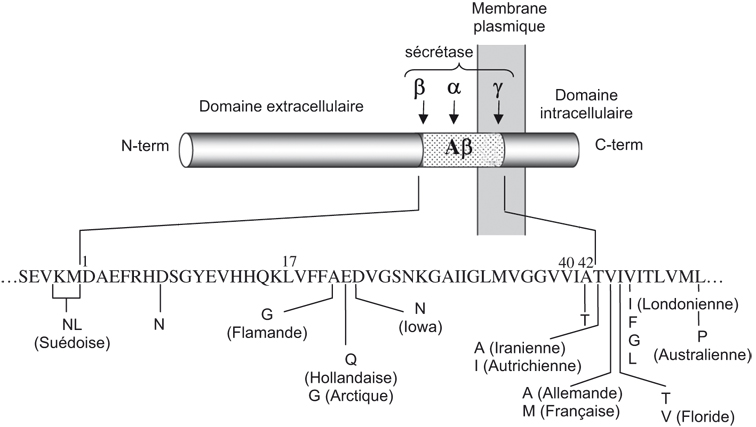
Le précurseur du peptide β-amyloïde (βAPP) est une protéine transmembranaire de 695 à 770 acides aminés. Le peptide Aβ résulte de l'action combinée de deux activités protéolytiques distinctes, la β-sécrétase et la γsécrétase, qui libèrent respectivement les extrémités N- et C-terminales du peptide (figure 2.2). C'est la voie dite « amyloïdogénique » de maturation de la βAPP (Checler, 1995). Il faut signaler ici un aspect important de ce métabolisme : la coupure par la γ-sécrétase conditionne la nature de l'extrémité C-terminale du peptide Aβ qui peut être de 40 (Aβ40) ou de 42 (Aβ42) acides aminés. Ceci n'est pas anodin puisque les formes de 42 acides aminés s'agrègent plus facilement (Burdick et coll., 1997) et sont généralement augmentées de manière sélective dans la maladie (pour revue voir : Selkoe, 2001). Récemment, une coupure additionnelle a été décrite (coupure ε) qui intervient en aval du site de la γ-sécrétase (Passer et coll., 2000 ; Gu et coll., 2001 ; Sastre et coll., 2001 ; Weidemann et coll., 2002) et libère un fragment (ICD ou AICD pour βAPP IntraCellular Domain) qui jouerait le rôle d'un facteur de transcription (Cao et Südhof, 2001 ; Baek et coll., 2002 ; Pardossi-Piquard et coll., 2005 ; Alves da Costa et coll., 2006). D'autre part, une coupure alternative intervient au milieu de la séquence Aβ par une activité α-sécrétase (figure 2.2) qui est responsable de la voie de maturation dite « non amyloïdogénique » (Checler, 1995). Non seulement cette coupure prévient la production de peptide Aβ mais aussi libère un fragment sécrété appelé sAPPα qui est trophique et neuroprotecteur (pour revue voir : Mattson, 1997).
). C'est la voie dite « amyloïdogénique » de maturation de la βAPP (Checler, 1995). Il faut signaler ici un aspect important de ce métabolisme : la coupure par la γ-sécrétase conditionne la nature de l'extrémité C-terminale du peptide Aβ qui peut être de 40 (Aβ40) ou de 42 (Aβ42) acides aminés. Ceci n'est pas anodin puisque les formes de 42 acides aminés s'agrègent plus facilement (Burdick et coll., 1997) et sont généralement augmentées de manière sélective dans la maladie (pour revue voir : Selkoe, 2001). Récemment, une coupure additionnelle a été décrite (coupure ε) qui intervient en aval du site de la γ-sécrétase (Passer et coll., 2000 ; Gu et coll., 2001 ; Sastre et coll., 2001 ; Weidemann et coll., 2002) et libère un fragment (ICD ou AICD pour βAPP IntraCellular Domain) qui jouerait le rôle d'un facteur de transcription (Cao et Südhof, 2001 ; Baek et coll., 2002 ; Pardossi-Piquard et coll., 2005 ; Alves da Costa et coll., 2006). D'autre part, une coupure alternative intervient au milieu de la séquence Aβ par une activité α-sécrétase (figure 2.2) qui est responsable de la voie de maturation dite « non amyloïdogénique » (Checler, 1995). Non seulement cette coupure prévient la production de peptide Aβ mais aussi libère un fragment sécrété appelé sAPPα qui est trophique et neuroprotecteur (pour revue voir : Mattson, 1997). | Figure 2.2 Maturation physiopathologique de la βAPP et mutations responsables de certaines formes familiales de maladie d'Alzheimer |
Maturation amyloïdogénique
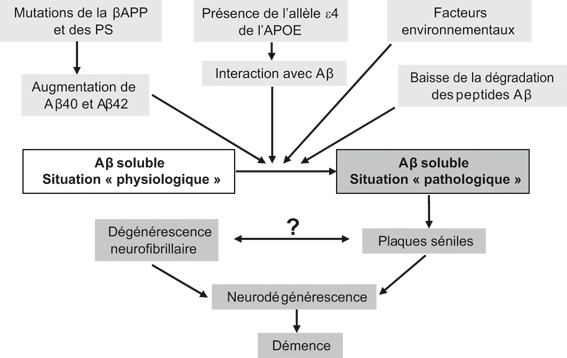
En préalable, il convient de souligner que la voie amyloïdogénique conduit à la production de peptide Aβ mais n'a pas de connotation pathogène systématique puisque le peptide peut ne pas s'agréger sous forme amyloïde. En effet, il est maintenant établi que le peptide Aβ est un produit de maturation physiologique de la βAPP (Haass et coll., 1992 ; Shoji et coll., 1992 ; Busciglio et coll., 1993). C'est la dérégulation de cette production, conduisant soit à une exacerbation de la production de peptide Aβ, soit à la production de catabolites toxiques qui est associée à la pathologie. Mieux comprendre les dérèglements qui font évoluer la cellule d'une situation physiologique vers une situation pathologique constitue donc un des principaux challenges de la recherche actuelle sur la maladie d'Alzheimer. Comme indiqué ci-dessus, certaines mutations de la βAPP (figure 2.2) peuvent rendre compte dans quelques cas rares de cette perturbation physiologique et de ses conséquences. Bien d'autres facteurs peuvent affecter cette production comme le montre la figure 2.3. Un autre challenge a consisté à identifier et caractériser les enzymes « pathogènes » c'est-à-dire les β- et γ- sécrétases ainsi que l'enzyme « bénéfique », l'α-sécrétase. De plus, de nombreuses études ont cherché à mieux cerner les processus par lesquels le peptide Aβ est dégradé par la cellule.
; Shoji et coll., 1992 ; Busciglio et coll., 1993). C'est la dérégulation de cette production, conduisant soit à une exacerbation de la production de peptide Aβ, soit à la production de catabolites toxiques qui est associée à la pathologie. Mieux comprendre les dérèglements qui font évoluer la cellule d'une situation physiologique vers une situation pathologique constitue donc un des principaux challenges de la recherche actuelle sur la maladie d'Alzheimer. Comme indiqué ci-dessus, certaines mutations de la βAPP (figure 2.2) peuvent rendre compte dans quelques cas rares de cette perturbation physiologique et de ses conséquences. Bien d'autres facteurs peuvent affecter cette production comme le montre la figure 2.3. Un autre challenge a consisté à identifier et caractériser les enzymes « pathogènes » c'est-à-dire les β- et γ- sécrétases ainsi que l'enzyme « bénéfique », l'α-sécrétase. De plus, de nombreuses études ont cherché à mieux cerner les processus par lesquels le peptide Aβ est dégradé par la cellule.
β-sécrétase
La β-sécrétase est l'enzyme qui libère l'extrémité N-terminale du peptide amyloïde. La nature de cette enzyme est maintenant consensuelle. Il s'agit d'une protéase acide purifiée et caractérisée simultanément par 5 laboratoires, appelée BACE1 (βAPP Cleaving Enzyme 1) ou memapsin 2 (Hussain et coll., 1999 ; Sinha et coll., 1999 ; Yan et coll., 1999 ; Lin et coll., 2000 ; Vassar, 2001). BACE1 présente un homologue appelé BACE2 qui semble peu abondant au niveau cérébral et qui ne contribue pas ou peu à la production de peptide Aβ. En effet, l'invalidation du gène codant pour BACE1 seul suffit à bloquer presque totalement la production de peptide Aβ (Cai et coll., 2001 ; Luo et coll., 2001). De manière intéressante, les souris invalidées pour BACE1 sont viables et fertiles (Luo et coll., 2001 ; Roberds et coll., 2001). Cette observation importante indique que BACE1 assure une activité enzymatique (qu'elle soit ou non confinée au clivage de l'APP), qui n'a pas de rôle majeur dans des fonctions vitales et de reproduction chez l'animal ou que ces fonctions peuvent être assurées, chez l'animal knock-out, par d'autres enzymes. Des résultats récents indiquent que la βAPP n'est pas le seul substrat de BACE1 qui peut notamment hydrolyser l'α2,6-sialyltransférase, in vitro et in vivo (Kitazume et coll., 2003 et 2005) ainsi que la neureguline (Willem et coll., 2006). L'ensemble de ces résultats semble indiquer qu'une stratégie thérapeutique visant à bloquer la β-sécrétase ne se heurte pas, a priori, à des effets indésirables insurmontables qui seraient directement liés à l'inhibition de l'enzyme.
; Sinha et coll., 1999 ; Yan et coll., 1999 ; Lin et coll., 2000 ; Vassar, 2001). BACE1 présente un homologue appelé BACE2 qui semble peu abondant au niveau cérébral et qui ne contribue pas ou peu à la production de peptide Aβ. En effet, l'invalidation du gène codant pour BACE1 seul suffit à bloquer presque totalement la production de peptide Aβ (Cai et coll., 2001 ; Luo et coll., 2001). De manière intéressante, les souris invalidées pour BACE1 sont viables et fertiles (Luo et coll., 2001 ; Roberds et coll., 2001). Cette observation importante indique que BACE1 assure une activité enzymatique (qu'elle soit ou non confinée au clivage de l'APP), qui n'a pas de rôle majeur dans des fonctions vitales et de reproduction chez l'animal ou que ces fonctions peuvent être assurées, chez l'animal knock-out, par d'autres enzymes. Des résultats récents indiquent que la βAPP n'est pas le seul substrat de BACE1 qui peut notamment hydrolyser l'α2,6-sialyltransférase, in vitro et in vivo (Kitazume et coll., 2003 et 2005) ainsi que la neureguline (Willem et coll., 2006). L'ensemble de ces résultats semble indiquer qu'une stratégie thérapeutique visant à bloquer la β-sécrétase ne se heurte pas, a priori, à des effets indésirables insurmontables qui seraient directement liés à l'inhibition de l'enzyme.γ-sécrétase
La γ-sécrétase est l'enzyme qui libère l'extrémité C-terminale des peptides amyloïdes, engendrant les couples Aβ40/AICDC59 et Aβ42/AICDC57 (figure 2.4). La simple observation de la topologie de ces hydrolyses montre que la γ-sécrétase est une protéase atypique puisqu'elle clive la βAPP au niveau de son insertion dans la membrane, c'est-à-dire dans un milieu hydrophobe a priori hostile au processus catalytique requérant une molécule d'eau. Il existe un autre clivage (coupure ε) intervenant en aval du site γ-sécrétase, proche du feuillet interne de la membrane, qui libère l'AICDC50 (figure 2.4). Le fait que les coupures aux sites γ- et ε- de la βAPP soient dues à la même activité protéolytique est encore discuté. En effet, alors que certains auteurs suggèrent que ces deux coupures sont dues à l'activité γ-sécrétase dépendante des présénilines (Gu et coll., 2001 ; Sastre et coll., 2001 ; Weidemann et coll., 2002), des études récentes indiquent qu'il est possible de discriminer les deux types par une approche mutationnelle de la βAPP ou pharmacologique grâce à des inhibiteurs de la γ-sécrétase (Levitan et coll., 1996 ; Zhang et coll., 2000a ; Chen et coll., 2002 ; Moelhmann et coll., 2002).
). La simple observation de la topologie de ces hydrolyses montre que la γ-sécrétase est une protéase atypique puisqu'elle clive la βAPP au niveau de son insertion dans la membrane, c'est-à-dire dans un milieu hydrophobe a priori hostile au processus catalytique requérant une molécule d'eau. Il existe un autre clivage (coupure ε) intervenant en aval du site γ-sécrétase, proche du feuillet interne de la membrane, qui libère l'AICDC50 (figure 2.4). Le fait que les coupures aux sites γ- et ε- de la βAPP soient dues à la même activité protéolytique est encore discuté. En effet, alors que certains auteurs suggèrent que ces deux coupures sont dues à l'activité γ-sécrétase dépendante des présénilines (Gu et coll., 2001 ; Sastre et coll., 2001 ; Weidemann et coll., 2002), des études récentes indiquent qu'il est possible de discriminer les deux types par une approche mutationnelle de la βAPP ou pharmacologique grâce à des inhibiteurs de la γ-sécrétase (Levitan et coll., 1996 ; Zhang et coll., 2000a ; Chen et coll., 2002 ; Moelhmann et coll., 2002).

De nombreuses études suggèrent que les présénilines 1 et 2 (PS1 et PS2), dont les mutations sont responsables de la majorité des formes familiales de maladie d'Alzheimer, sont elles-mêmes porteuses de l'activité γ-sécrétase. L'observation empirique que les mutations portées par les PS se traduisent toujours par une modulation des taux et de la nature même du peptide Aβ formé, avec une incidence particulière sur la production exacerbée de Aβ42 pathogène (pour revue voir : Checler, 1999a) l'a initialement laissé supposer. D'autre part, il est intéressant de noter que l'invalidation du gène codant la PS1 diminue drastiquement la production de peptides Aβ et que cette dernière est virtuellement abolie quand PS1 et PS2 sont déplétées (Herreman et coll., 2000 ; Zhang et coll., 2000b). Il faut souligner ici que plusieurs études ont démontré la présence d'une activité γ-sécrétase indépendante des présénilines.
) l'a initialement laissé supposer. D'autre part, il est intéressant de noter que l'invalidation du gène codant la PS1 diminue drastiquement la production de peptides Aβ et que cette dernière est virtuellement abolie quand PS1 et PS2 sont déplétées (Herreman et coll., 2000 ; Zhang et coll., 2000b). Il faut souligner ici que plusieurs études ont démontré la présence d'une activité γ-sécrétase indépendante des présénilines.Des études récentes ont établi qu'il ne fallait pas parler de γ-sécrétase mais que le terme de complexe γ-sécrétase dépendant des présénilines était vraisemblablement plus approprié. En effet, l'activité γ-sécrétase dépendante des PS apparaît portée par un complexe multiprotéique de haut poids moléculaire impliquant au moins trois autres protéines, la nicastrine (NCT), Aph1 (Anterior pharynx defective 1 homolog) and Pen-2 (Presenilin enhancer 2 homolog) (Goutte et coll., 2000 ; Yu et coll., 2000 ; Francis et coll., 2002). Les présénilines sont des protéines transmembranaires (pour revue voir : Checler, 1999b) (figure 2.5) qui subissent une coupure par une « présénilinase ». Les fragments N- et C-terminaux, issus de cette coupure, interagissent stochiométriquement pour former l'entité biologiquement active (Thinakaran et coll., 1996). La NCT, Aph-1 et Pen-2 sont aussi des protéines présentant un (NCT) ou plusieurs (Aph-1 et Pen-2) domaines transmembranaires (figure 2.5).
; Yu et coll., 2000 ; Francis et coll., 2002). Les présénilines sont des protéines transmembranaires (pour revue voir : Checler, 1999b) (figure 2.5) qui subissent une coupure par une « présénilinase ». Les fragments N- et C-terminaux, issus de cette coupure, interagissent stochiométriquement pour former l'entité biologiquement active (Thinakaran et coll., 1996). La NCT, Aph-1 et Pen-2 sont aussi des protéines présentant un (NCT) ou plusieurs (Aph-1 et Pen-2) domaines transmembranaires (figure 2.5).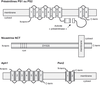
Même si l'hypothèse d'autres contributeurs protéiques au complexe γ-sécrétase ne peut être totalement écartée (il existe notamment des régulateurs de ce complexe comme TMP21 ; Chen et coll., 2006), il est notable que l'absence d'une seule des quatre protéines décrites ci-dessus affecte massivement la production de peptide Aβ (Edbauer et coll., 2003 ; Takasugi et coll., 2003). Ceci est dû à une interaction physique intime de ces protéines. En effet, la formation du complexe γ-sécrétase dépendant des PS est régie par une séquence d'événements maintenant bien caractérisée (figure 2.6). Ainsi, la NCT forme tout d'abord un sous-complexe avec Aph-1 vraisemblablement très tôt dans le réticulum endoplasmique puisque Aph-1 interagit avec les formes déglycosylées de NCT (Hu et Fortini, 2003 ; La Voie et coll., 2003). Après sa formation, ce sous-complexe interagit avec la PS1 ou la PS2 (Lee et coll., 2002 ; Gu et coll., 2003). Enfin, Pen-2 s'associe au complexe et promeut vraisemblablement l'hydrolyse des PS, conduisant finalement au complexe biologiquement actif (Luo et coll., 2003 ; Takasugi et coll., 2003).
), il est notable que l'absence d'une seule des quatre protéines décrites ci-dessus affecte massivement la production de peptide Aβ (Edbauer et coll., 2003 ; Takasugi et coll., 2003). Ceci est dû à une interaction physique intime de ces protéines. En effet, la formation du complexe γ-sécrétase dépendant des PS est régie par une séquence d'événements maintenant bien caractérisée (figure 2.6). Ainsi, la NCT forme tout d'abord un sous-complexe avec Aph-1 vraisemblablement très tôt dans le réticulum endoplasmique puisque Aph-1 interagit avec les formes déglycosylées de NCT (Hu et Fortini, 2003 ; La Voie et coll., 2003). Après sa formation, ce sous-complexe interagit avec la PS1 ou la PS2 (Lee et coll., 2002 ; Gu et coll., 2003). Enfin, Pen-2 s'associe au complexe et promeut vraisemblablement l'hydrolyse des PS, conduisant finalement au complexe biologiquement actif (Luo et coll., 2003 ; Takasugi et coll., 2003).

Le schéma ci-dessus ne traduit pas totalement la complexité de cet assemblage moléculaire. Parce qu'il existe deux présénilines, trois homologues de Aph-1 (Aph-1a, Aph-1b et Aph-1c) (Francis et coll., 2002 ; Goutte et coll., 2002 ; Ma et coll., 2005), et deux isoformes de Aph-1a (Aph-1aL et Aph1aS) (Francis et coll., 2002 ; Goutte et coll., 2002 ; Gu et coll., 2003), différents types de complexes contenant diverses combinaisons de protéines peuvent être envisagés. Cette hypothèse a été corroborée par des travaux récents identifiant différents complexes de composition protéique spécifique (Hébert et coll., 2004 ; Shirotani et coll., 2004). Cependant, la régulation de la composition de ces complexes demeure mal comprise et il n'est pas sûr que le processus de formation du complexe γ-sécrétase dépendant des PS biologiquement actif comporte une séquence invariable d'événements. En effet, par des expériences de dissociation, par des détergents, du complexe γ-sécrétase biologiquement actif, il a été montré qu'il existait plusieurs complexes majeurs et mineurs de compositions différentes (Fraering et coll., 2004). Les complexes γ-sécrétase distincts pourraient avoir des fonctions différentes liées à la capacité d'hydrolyse de substrats spécifiques. Ces observations apportent naturellement un degré supplémentaire de difficulté dans l'optique d'une stratégie visant à bloquer l'activité γ-sécrétase dépendante des présénilines puisqu'il faudrait idéalement développer des inhibiteurs spécifiques à chacun des complexes.
; Goutte et coll., 2002 ; Ma et coll., 2005), et deux isoformes de Aph-1a (Aph-1aL et Aph1aS) (Francis et coll., 2002 ; Goutte et coll., 2002 ; Gu et coll., 2003), différents types de complexes contenant diverses combinaisons de protéines peuvent être envisagés. Cette hypothèse a été corroborée par des travaux récents identifiant différents complexes de composition protéique spécifique (Hébert et coll., 2004 ; Shirotani et coll., 2004). Cependant, la régulation de la composition de ces complexes demeure mal comprise et il n'est pas sûr que le processus de formation du complexe γ-sécrétase dépendant des PS biologiquement actif comporte une séquence invariable d'événements. En effet, par des expériences de dissociation, par des détergents, du complexe γ-sécrétase biologiquement actif, il a été montré qu'il existait plusieurs complexes majeurs et mineurs de compositions différentes (Fraering et coll., 2004). Les complexes γ-sécrétase distincts pourraient avoir des fonctions différentes liées à la capacité d'hydrolyse de substrats spécifiques. Ces observations apportent naturellement un degré supplémentaire de difficulté dans l'optique d'une stratégie visant à bloquer l'activité γ-sécrétase dépendante des présénilines puisqu'il faudrait idéalement développer des inhibiteurs spécifiques à chacun des complexes.α-sécrétase
La voie α-sécrétase est non amyloïdogénique puisque la coupure par cette enzyme intervient au milieu de la séquence Aβ portée par la βAPP (figure 2.2). Cette coupure engendre un produit N-terminal de la βAPP qui est sécrété, l'APPα. L'APPα a son propre spectre biologique et il peut notamment protéger la cellule de la toxicité médiée par le peptide Aβ (Mattson, 1997). Il existe deux voies α-sécrétase distinctes : l'une constitutive et l'autre régulée (pour revue voir : Checler, 1995). Brièvement, la voie sécrétoire régulée est sous le contrôle de la protéine kinase C (PKC) et elle conduit de façon concomitante à la réduction de la production de peptide Aβ, en accord avec l'hypothèse selon laquelle les productions d'APPα et d'Aβ ne seraient pas mutuellement exclusives. Ceci a été validé in vivo puisque diverses études ont montré que la stimulation de la voie PKC conduisait à la baisse de peptide Aβ chez des souris transgéniques porteuses du gène muté de l'APP comportant une séquence humanisée de l'Aβ (Savage et coll., 1998). La voie α-sécrétase est aussi contrôlée par la protéine kinase A (Checler, 1995 ; Marambaud et coll., 1998). Il existe plusieurs α-sécrétases qui sont toutes des métalloprotéases qui appartiennent à la famille des disintégrines. ADAM10 (A Disintegrin And Metalloprotease 10) contribue aux deux voies sécrétoires, constitutive (Lammich et coll., 1999 ; Lopez-Perez et coll., 2001) et régulée, alors qu'ADAM17 (appelée aussi TACE, pour Tumor necrosis Alpha Converting Enzyme) est essentiellement responsable de la voie de sécrétion régulée (Buxbaum et coll., 1998). D'autres études, plus discutées, ont suggéré qu'ADAM9 pourrait aussi participer à la maturation physiologique de la βAPP et jouer le rôle d'α-sécrétase (Koike et coll., 1999 ; Hotoda et coll., 2002). Il est intéressant de noter qu'une étude récente a établi que la manipulation génétique d'ADAM10, se traduisant par une augmentation de son expression, réduisait la production de peptide Aβ et avait des effets bénéfiques sur les troubles cognitifs associés à ce peptide (Postina et coll., 2004), ce qui supporte l'hypothèse d'une stratégie thérapeutique visant à augmenter l'expression de l'α-sécrétase et donc à réduire les taux de peptide Aβ.
). Cette coupure engendre un produit N-terminal de la βAPP qui est sécrété, l'APPα. L'APPα a son propre spectre biologique et il peut notamment protéger la cellule de la toxicité médiée par le peptide Aβ (Mattson, 1997). Il existe deux voies α-sécrétase distinctes : l'une constitutive et l'autre régulée (pour revue voir : Checler, 1995). Brièvement, la voie sécrétoire régulée est sous le contrôle de la protéine kinase C (PKC) et elle conduit de façon concomitante à la réduction de la production de peptide Aβ, en accord avec l'hypothèse selon laquelle les productions d'APPα et d'Aβ ne seraient pas mutuellement exclusives. Ceci a été validé in vivo puisque diverses études ont montré que la stimulation de la voie PKC conduisait à la baisse de peptide Aβ chez des souris transgéniques porteuses du gène muté de l'APP comportant une séquence humanisée de l'Aβ (Savage et coll., 1998). La voie α-sécrétase est aussi contrôlée par la protéine kinase A (Checler, 1995 ; Marambaud et coll., 1998). Il existe plusieurs α-sécrétases qui sont toutes des métalloprotéases qui appartiennent à la famille des disintégrines. ADAM10 (A Disintegrin And Metalloprotease 10) contribue aux deux voies sécrétoires, constitutive (Lammich et coll., 1999 ; Lopez-Perez et coll., 2001) et régulée, alors qu'ADAM17 (appelée aussi TACE, pour Tumor necrosis Alpha Converting Enzyme) est essentiellement responsable de la voie de sécrétion régulée (Buxbaum et coll., 1998). D'autres études, plus discutées, ont suggéré qu'ADAM9 pourrait aussi participer à la maturation physiologique de la βAPP et jouer le rôle d'α-sécrétase (Koike et coll., 1999 ; Hotoda et coll., 2002). Il est intéressant de noter qu'une étude récente a établi que la manipulation génétique d'ADAM10, se traduisant par une augmentation de son expression, réduisait la production de peptide Aβ et avait des effets bénéfiques sur les troubles cognitifs associés à ce peptide (Postina et coll., 2004), ce qui supporte l'hypothèse d'une stratégie thérapeutique visant à augmenter l'expression de l'α-sécrétase et donc à réduire les taux de peptide Aβ.Dégradation du peptide Aβ
Les taux endogènes de peptides Aβ sont régis par la balance entre les processus de formation du peptide et ceux de sa dégradation. Il n'existe aucune étude montrant que les processus de formation des peptides amyloïdes sont altérés dans les formes sporadiques de la maladie d'Alzheimer et, notamment, aucun travail n'a établi que l'activité des β- et γ-sécrétases était augmentée. On admet généralement que les modifications se traduisant par l'augmentation des niveaux de peptide Aβ sont principalement post-traductionnelles. Les processus de dégradation du peptide sont donc particulièrement importants pour éviter son accumulation. Dans la majorité des travaux, les protéases impliquées ont été identifiées en examinant l'influence de leur inactivation ou de leur surexpression sur les taux de peptide Aβ, le nombre de plaques séniles et les processus cognitifs chez les animaux « Alzheimerisés ». Ces études ont conduit à identifier la néprilysine (NEP ; Hama et coll., 2001 ; Iwata et coll., 2001 ; Hauss-Wegrzyniak et Wenk, 2002 ; Leissring et coll., 2003 ; Marr et coll., 2003 ; Hama et coll., 2004 ; Marr et coll., 2004), l'enzyme de conversion de l'endothéline (ECE ; Eckman et coll., 2003) et l'enzyme de dégradation de l'insuline (IDE ; Farris et coll., 2003). Très récemment, il a été établi que les AICD modulaient au niveau transcriptionnel l'expression et l'activité de NEP mais pas celles d'ECE et IDE. Ces résultats suggèrent un lien entre activité sécrétase et enzyme de dégradation de NEP et montrent que l'on peut moduler l'activité de la γ-sécrétase sans que cela se traduise systématiquement par une augmentation de peptide Aβ.
; Iwata et coll., 2001 ; Hauss-Wegrzyniak et Wenk, 2002 ; Leissring et coll., 2003 ; Marr et coll., 2003 ; Hama et coll., 2004 ; Marr et coll., 2004), l'enzyme de conversion de l'endothéline (ECE ; Eckman et coll., 2003) et l'enzyme de dégradation de l'insuline (IDE ; Farris et coll., 2003). Très récemment, il a été établi que les AICD modulaient au niveau transcriptionnel l'expression et l'activité de NEP mais pas celles d'ECE et IDE. Ces résultats suggèrent un lien entre activité sécrétase et enzyme de dégradation de NEP et montrent que l'on peut moduler l'activité de la γ-sécrétase sans que cela se traduise systématiquement par une augmentation de peptide Aβ.En conclusion,
même si la preuve définitive de l'implication du peptide amyloïde en tant que premier déterminant étiologique de la maladie d'Alzheimer reste à apporter, le peptide amyloïde demeure au centre de la physiopathologie. L'hypothèse de la cascade amyloïde présente en outre l'avantage de cadrer des cibles très en amont de la cascade pathologique (enzymes de production, enzymes de dégradation). À terme, interférer avec ces cibles pourrait obérer le processus neurodégénératif et tous les effets délétères associés à la production de peptide amyloïde. Il est possible que l'immunisation contre le peptide Aβ qui réduit de façon importante son accumulation intracérébrale permette rapidement de mieux comprendre son rôle physiopathologique. D'autre part, la cascade prédit une relation directe entre l'accumulation extracellulaire de peptide Aβ et les accumulations intracellulaires de protéine Tau. De nombreuses hypothèses ont été formulées mais aucune n'a permis, à ce jour, de reconstituer expérimentalement des dégénérescences neurofibrillaires à partir d'une surexpression de peptide Aβ. Enfin, l'hypothèse de la cascade telle qu'elle a été initialement formulée faisait jouer un rôle essentiel au rôle toxique du peptide Aβ dans sa forme native : des données récentes suggèrent que le peptide s'assemble rapidement sous forme d'oligomères. Ces oligomères et particulièrement les trimères inhibent expérimentalement la potentialisation à long terme telle qu'elle est étudiée sur des tranches d'hippocampe. Le peptide Aβ pourrait donc avoir un effet fonctionnel avant de provoquer des lésions visibles.
Bibliographie
[1] alves da costa c, sunyach c, pardossi-piquard r, sevalle j, vincent b. Presenilin-dependent γ-secretase-mediated control of p53-associated cell death in Alzheimer’s disease.
J Neurosci. 2006;
26:6377- 6385
[2]alzheimer’s disease collaborative group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families.
Nature Genetics. 1996;
11:219- 222
[3] baek sh, ohgi ka, rose dw, koo eh, glass ck. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein.
Cell. 2002;
110:55- 67
[4] burdick d, kosmoski j, knauer mf, glabe cg. Preferential adsorption, internalization and resistance to degradation of the major isoform of the Alzheimer’s amyloid peptide, Aβ1-42, in differentiated PC12.
Brain Res. 1997;
746:275- 284
[5] busciglio j, gabuzda dh, matsudaira p, yankner ba. Generation of β amyloid in the secretory pathway in neuronal and non neuronal cells.
Proc Natl Acad Sci USA. 1993;
90:2092- 2096
[6] buxbaum jd, liu kn, luo y, slack jl, stocking kl. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor.
J Biol Chem. 1998;
273:27765- 27767
[7] cai h, wang y, mccarthy d, wen h, borchelt dr. BACE1 is the major β-secretase for generation of Aβ peptides by neurons.
Nat Neurosci. 2001;
4:233- 234
[8] cao x, südhof tc. A transcriptively active complex of APP with Fe65 and histone acetyltransferase tip60.
Science. 2001;
293:115- 120
[9] checler f. Processing of the β-amyloid precursor protein and its regulation in Alzheimer’s disease.
J Neurochem. 1995;
65:1431- 1444
[10] checler f. Presenilins: multifunctional proteins involved in Alzheimer’s disease pathology.
Iubmb LIFE. 1999a;
48:33- 39
[11] checler f. Presenilins: Structural aspects and post-translational events.
Mol Neurobiol. 1999b;
19:255- 265
[12] chen f, gu y, hasegawa h, ruan x, arawaka s. Presenilin 1 mutations activate γ-42-secretase but reciprocally inhibit µ-secretase cleavage of APP and S3-cleavage of Notch.
J Biol Chem. 2002;
277:36521- 36526
[13] chen f, hasegawa h, schmitt-ulms g, kawarai t, bohm c. TMP21 is a presenilin complex component that modulates γ- but not ε-secretase activities.
Nature. 2006;
440:1208- 1212
[14] eckman ea, watson m, marlow l, sambamurti k, eckman cb. Alzheimer’s disease β-amyloid peptide (Ab) is increased in mice deficient in endothelin-converting enzyme.
J Biol Chem. 2003;
278:2081- 2084
[15] edbauer d, winkler e, regula jt, pesold b, steiner h. Reconstitution of γ-secretase activity.
Nat Cell Biol. 2003;
5:486- 488
[16] farris w, mansourian s, chang y, lindsley l, eckman ea. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo.
Proc Natl Acad Sci USA. 2003;
100:4162- 4167
[17] fraering pc, la voie mj, ye w, ostaszewski bl, kimberly wt. Detergent-dependent dissociation of active g-secretase reveals an interaction between pen-2 and PS1-NTF and offers a model for subunit organization within the complex.
Biochemistry. 2004;
43:323- 333
[18] francis r, mcgrath g, zhang j, ruddy da, sym m. Aph-1 and pen2 are required for notch pathway signaling, γ-secretase cleavage of βAPP and presenilin protein accumulation.
Dev cell. 2002;
3:85- 97
[19] glenner gg, wong cw. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein.
BBRC. 1984a;
120:885- 890
[20] glenner gg, wong cw. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein.
BBRC. 1984b;
122:1131- 1135
[21] goldgaber d, lerman mi, mcbride ow, saffiotti u, gajdusek dc. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease.
Science. 1987;
235:887- 880
[22] goutte c, hepler w, mickey k, priess j. aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling.
Development. 2000;
127:2481- 2492
[23] goutte c, tsunozaki m, hale va, priess jr. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos.
Proc Natl Acad Sci USA. 2002;
99:775- 779
[24] gu y, chen f, sanjo n, kawarai t, hasegawa h. APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin-nicastrin complexes.
J Biol Chem. 2003;
278:7374- 7380
[25] gu y, misonou h, sato t, dohmae n, takio k. Distinct intramembrane cleavages of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch.
J Biol Chem. 2001;
276:35235- 35238
[26] haass c, schlossmacher mg, hung ay, vigo-pelfrey c, mellon a. Amyloïd β-peptide is produced by cultured cells during normal metabolism.
Nature. 1992;
359:322- 325
[27] hama e, shirotani k, masumoto h, sekine-aizawa y, aizawa h. Clearance of extracellular and cell-associated amyloid b peptide through viral expression of neprilysin in primary neurons.
J Biochem. 2001;
130:721- 726
[28] hama e, shirotani k, iwata a, saido tc. Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid β peptide clearance in primary neurons.
J Biol Chem. 2004;
279:30259- 30264
[29] hauss-wegrzyniak b, wenk gl. Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: the role of ascorbic acid in the vehicle.
Neurosci Lett. 2002;
322:75- 78
[30] hébert ss, serneels l, dejaegere t, horré k, dabrowski m. Coordinated and widespread expression of γ-secretase in vivo: evidence for size and molecular heterogeneity.
Neurobiol of Dis. 2004;
17:260- 272
[31] herreman a, serneels l, annaert w, collen d, schoonjans l. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells.
Nat Cell Biol. 2000;
2:461- 462
[32] hotoda n, koike h, sasagawa n, ishiura s. A secreted form of ADAM9 has an α-secretase activity for APP.
Biochem Biophys Res Commun. 2002;
293:800- 805
[33] hu y, fortini me. Different cofactor activities in γ-secretase assembly: evidence for a nicastrin-Aph-1 subcomplex.
J Cell Biol. 2003;
161:685- 690
[34] hussain i, powell d, howlett dr, tew dg, meek td. Identification of a novel aspartic protease (Asp2) as β-secretase.
Mol Cell Neurosci. 1999;
14:419- 427
[35] iwata n, tsubuki s, takaki y, shirotani k, lu b. Metabolic regulation of brain Aβ by neprilysin.
Science. 2001;
292:1550- 1552
[36] kang j, lemaire hg, unterbeck a, salbaum jm, masters cl. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor.
Nature. 1987;
325:733- 736
[37] kitazume s, tachida y, oka r, kotani n, ogawa k. Characterization of α2,6-sialyltransferase cleavage by Alzheimer’s β-secretase (BACE1).
J Biol Chem. 2003;
278:14865- 14871
[38] kitazume s, nakagawa k, oka r, tachida y, ogawa k. In vivo cleavage of α2,6-sialyltransferase by Alzheimer’s β-secretase.
J Biol Chem. 2005;
280:8589- 8595
[39] koike h, tomioka s, sorimachi h, saido tc, maruyama k. Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein.
Biochem J. 1999;
343:371- 375
[40] la voie mj, fraering pc, ostaszewski bl, yew , kimberly wt. Assembly of the γ-secretase complex involves early formation of an intermediate sub-complex of Aph-1 and nicastrin.
J Biol Chem. 2003;
278:37213- 37222
[41] lammich s, kojro e, postina r, gilbert s, pfeiffer r. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease.
Proc Natl Acad Sci USA. 1999;
96:3922- 3927
[42] lee s-f, shah s, li h, yu c, yu g. Mammalian APH-1 interacts with presenilin and nicastrin, and is required for intramembrane proteolysis of APP and Notch.
J Biol Chem. 2002;
277:45013- 45019
[43] leissring ma, farris w, chang ay, walsh dm, wu x. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology and premature death.
Neuron. 2003;
40:1087- 1093
[44] levitan d, doyle tg, brousseau d, lee mk, thinakaran g. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans.
Proc Natl Acad Sci USA. 1996;
93:14940- 14944
[45] levy-lahad e, wasco w, poorkaj p, romano dm, oshima j. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus.
Science. 1995;
269:973- 977
[46] lin x, koelsch g, wu s, downs d, dashti a. Human aspartyl protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein.
Proc Natl Acad Sci USA. 2000;
97:1456- 1460
[47] lopez-perez e, zhang y, franck sj, creemers j, seidah n. Constitutive α-secretase cleavage of the β-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the prohormone convertase 7 (PC7) and the disintegrin metalloprotease ADAM10.
J Neurochem. 2001;
76:1532- 1539
[48] luo y, bolon b, kahn s, bennett bd, babu-khan s. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation.
Nat Neurosci. 2001;
4:231- 232
[49] luo wj, wang h, li h, kim bs, shah s. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1.
J Biol Chem. 2003;
278:7850- 7854
[50] ma g, li t, price d, wong pc. APH-1a is the principal mammalian APH-1 isoform present in γ-secretase complexes during embryonic development.
J Neurosci. 2005;
25:192- 198
[51] marambaud p, chevallier n, ancolio k, checler f. Post-transcriptional contribution of a cAMP-dependent pathway to the formation of α-and β/γ-secretases-derived products of βAPP maturation in human cells expressing wild type and Swedish mutated βAPP.
Mol Medicine. 1998;
4:715- 723
[52] marr ra, rockenstein e, mukerjee a, kindy ms, hersh lb. Neprilysin gene transfert reduces human amyloid pathology in transgenic mice.
J Neurosci. 2003;
23:1292- 1296
[53] marr ra, guan h, rockenstein e, kindy m, gage fh. Neprilysin regulates amyloid β peptide levels.
J Mol Neurosci. 2004;
22:5- 11
[54] masters cl, simons g, weinman na, multhaup g, mc donald bl. Amyloid plaque core protein in Alzheimer’s Disease and Down Syndrome.
Proc Natl Acad Sci USA. 1985;
82:4245- 4249
[55] mattson mp. Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives.
Physiol Rev. 1997;
77:1081- 1132
[56] moelhmann t, winkler e, xia x, edbauer d, murrell j. Presenilin-1 mutations of leucine 166 equally affect the generation of the notch and APP intracellular domains independent of their effect on Aβ42 production.
Proc Natl Acad Sci USA. 2002;
99:8025- 8030
[57] mullan m, crawford f. Genetic and molecular advances in Alzheimer’s disease.
Trends in Neurosci. 1993;
16:398- 403
[58] pardossi-piquard r, petit a, kawarai t, sunyach c, alves da costa c. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP.
Neuron. 2005;
46:541- 554
[59] passer b, pellegrini l, russo c, siegel rm, lenardio mj. Generation of an apoptotic intracellular peptide by γ-secretase cleavage of Alzheimer’s amyloid β protein precursor.
J Alzheimer’s Disease. 2000;
2:289- 301
[60] postina r, schroeder a, dewachter i, bohl j, schmitt u. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer’s disease mouse model.
J Clin Invest. 2004;
113:1456- 1464
[61] roberds sl, anderson j, basi g, bienkowski mj, branstetter dg. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer’s disease therapeutics.
Hum Mol Gen. 2001;
10:1317- 1324
[62] rogaev ei, sherrington r, rogaeva ea, levesque g, ikeda m. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene.
Nature. 1995;
376:775- 778
[63] sastre m, steiner h, fuchs k, capell a, multhaup g. Presenilin dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch.
EMBO report. 2001;
2:835- 841
[64] savage m, trusko sp, howland ds, pinsker lr, mistrett as. Turnover of amyloid β-protein in mouse brain and acute reduction of its level by phorbol ester.
J Neurosci. 1998;
18:1743- 1752
[65] schellenberg gd. Genetic dissection of Alzheimer disease, a heterogeneous disorder.
Proc Natl Acad Sci USA. 1995;
92:8552- 8559
[66] selkoe dj. Alzheimer’s disease: Genes, proteins and therapy.
Physiol Rev. 2001;
81:741- 766
[67] sherrington r, rogaev ei, liang y, rogaeva ea, levesque g. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease.
Nature. 1995;
375:754- 760
[68] shirotani k, edbauer d, prokop s, haass c, steiner h. Identification of distinct γ-secretase complexes with different APH-1 variants.
J Biol Chem. 2004;
279:41340- 41345
[69] shoji m, golde te, ghiso j, cheung tt, estus s. Production of the Alzheimer amyloid β protein by normal proteolytic processing.
Science. 1992;
258:126- 129
[70] sinha s, anderson jp, barbour r, basi gs, caccavello r. Purification and cloning of amyloid precursor protein β-secretase from human brain.
Nature. 1999;
402:537- 540
[71] takasugi n, tomita t, hayashi i, tsuruoka m, niimura m. The role of presenilin cofactors in the γ-secretase complex.
Nature. 2003;
422:438- 441
[72] tanzi re, gusella jf, watkins pc, bruns gap, st george-hyslop p. Amyloid β protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus.
Science. 1987;
235:880- 884
[73] tanzi re, st george-hyslop p, gusella jf. Molecular genetics of Alzheimer disease amyloid.
J Biol Chem. 1991;
266:20579- 20582
[74] thinakaran g, borchelt dr, lee mk, slunt hh, spitzer l. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo.
Neuron. 1996;
17:181- 190
[75] vassar r. The β-secretase, BACE.
J Mol Neurosci. 2001;
17:157- 170
[76] weidemann a, eggert s, reinhard fbm, vogel m, paliga k. A novel ε-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with the Notch processing.
Biochemistry. 2002;
41:2825- 2835
[77] willem m, garratt an, novak b, citron m, kaufmann s. Control of peripheral nerve myelination by the beta-secretase BACE1.
Science. 2006;
314:664- 666
[78] yan r, bienkowski mj, shuck me, miao h, tory mc. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity.
Nature. 1999;
402:533- 537
[79] yu g, nishimura m, arawaka s, levitan d, zhang l. Nicastrin modulates presenilin-mediated notch/glp1 signal transduction and βAPP processing.
Nature. 2000;
407:48- 54
[80] zhang dm, levitan d, yu g, nishimura m, chen f. Mutation of the conserved N-terminal cysteine (Cys92) of human presenilin1 causes increased A beta secretion in mammalian cells but impaired Notch/lin-12 signalling in C. elegans.
Neuroport. 2000a;
11:3227- 3230
[81] zhang z, nadeau p, song w, donoviel d, yuan m. Presenilins are required for γ-secretase cleavage of βAPP and transmembrane cleavage of Notch.
Nat Cell Biol. 2000b;
2:463- 465
→ Aller vers SYNTHESE