Connaissances fondamentales
2007
| ANALYSE |
7-
Imagerie cérébrale1
Auparavant considérée comme un moyen d'exclure une pathologie curable lors de l'établissement du diagnostic de maladie d'Alzheimer, l'imagerie cérébrale s'avère aujourd'hui d'un grand intérêt d'un point de vue fondamental, car elle contribue à une meilleure compréhension de la physiopathologie de cette maladie. De plus, elle devrait devenir un outil d'importance sur le plan clinique, en particulier dans le diagnostic précoce et la prédiction de l'évolution des troubles (Scheltens et Korf, 2000 ; Devous, 2002 ; Bastos Leite et coll., 2004 ; Norfray et Provenzale, 2004 ; Zamrini et coll., 2004 ; pour revues sur l'ensemble de ces méthodes).
; Devous, 2002 ; Bastos Leite et coll., 2004 ; Norfray et Provenzale, 2004 ; Zamrini et coll., 2004 ; pour revues sur l'ensemble de ces méthodes).
; Devous, 2002 ; Bastos Leite et coll., 2004 ; Norfray et Provenzale, 2004 ; Zamrini et coll., 2004 ; pour revues sur l'ensemble de ces méthodes).Les lésions neuropathologiques de la maladie d'Alzheimer conduisent au dysfonctionnement et à la mort des neurones qui se traduisent au niveau macroscopique par une altération de l'activité cérébrale et une atrophie du cerveau. Ces altérations macroscopiques peuvent désormais être étudiées in vivo, grâce à la tomographie par émission de positons (TEP) et à l'imagerie par résonance magnétique (IRM). Les modifications fonctionnelles qui concernent surtout le cortex associatif postérieur contrastent avec les altérations structurales qui touchent en premier lieu la région hippocampique. Cette discordance entre les deux profils d'atteinte cérébrale suggère des hypothèses qui trouvent un support dans les études d'activation en IRM fonctionnelle (IRMf) et en TEP ainsi que dans les études en IRM du tenseur de diffusion (DTI pour Diffusion Tensor Imaging) qui s'intéressent à la connectivité entre les différentes structures. Ces études permettent également de mieux comprendre le rôle des altérations cérébrales dans l'apparition des troubles cognitifs et les mécanismes compensatoires, qui diffèrent selon le stade de la maladie. Enfin, grâce au développement de nouveaux marqueurs, il est maintenant possible de marquer in vivo les atteintes neuropathologiques de la maladie d'Alzheimer.
Profil d'atrophie dans la maladie d'Alzheimer
Le profil d'atrophie dans la maladie d'Alzheimer est maintenant bien décrit grâce à l'IRM anatomique. Cette technique d'imagerie cérébrale permet d'étudier les modifications de densité et de volume de substance grise, donc l'atrophie, à partir d'images anatomiques acquises en trois dimensions. Les premières études en IRM anatomique se sont focalisées sur le lobe temporal interne, cible privilégiée des dégénérescences neurofibrillaires et impliqué dans les troubles de la mémoire qui dominent le tableau clinique de la maladie d'Alzheimer. Grâce à une analyse visuelle (Scheltens et coll., 1997 ; Korf et coll., 2004) ou à la méthode des régions d'intérêt (ROIs) (Du et coll., 2001), qui consiste à tracer le contour de la région manuellement, l'atrophie de cette structure, et plus particulièrement de l'hippocampe et du cortex entorhinal, a été mise en évidence dans la maladie d'Alzheimer dès le stade léger (Scheltens et coll., 2002 ; Chételat et Baron, 2003 ; pour revues). L'altération structurale de cette région est déjà présente au stade pré-démentiel de MCI et elle semble distinguer, parmi les patients atteints de MCI (Mild Cognitive Impairement), ceux qui évoluent vers une maladie d'Alzheimer (« converteurs ») des « non converteurs » avec une efficacité variable selon les études (Mosconi et coll., 2007, pour revue).
; Korf et coll., 2004) ou à la méthode des régions d'intérêt (ROIs) (Du et coll., 2001), qui consiste à tracer le contour de la région manuellement, l'atrophie de cette structure, et plus particulièrement de l'hippocampe et du cortex entorhinal, a été mise en évidence dans la maladie d'Alzheimer dès le stade léger (Scheltens et coll., 2002 ; Chételat et Baron, 2003 ; pour revues). L'altération structurale de cette région est déjà présente au stade pré-démentiel de MCI et elle semble distinguer, parmi les patients atteints de MCI (Mild Cognitive Impairement), ceux qui évoluent vers une maladie d'Alzheimer (« converteurs ») des « non converteurs » avec une efficacité variable selon les études (Mosconi et coll., 2007, pour revue).La méthode d'analyse visuelle qui peut s'appuyer sur une échelle de taux d'atrophie en plusieurs points peut sembler facile à utiliser (Korf et coll., 2004), mais il s'agit d'une méthode subjective, qui demande de l'expertise et ne permet pas d'obtenir des données quantitatives du degré d'atrophie. La méthode des ROIs présente aussi des limites, dans la mesure où leur délimitation dépend de l'expertise de l'observateur. L'ensemble de ces méthodes ne permet pas d'étudier l'atrophie cérébrale de manière exhaustive et objective. Une méthode automatique et reproductible a donc été employée pour analyser l'ensemble du cerveau, voxel par voxel (Voxel Based Morphometry, VBM). Cette méthode, utilisée pour la première fois dans un groupe important de patients par Baron et coll. (2001), puis par de nombreux autres auteurs (Ohnishi et coll., 2001 ; Burton et coll., 2002 ; Frisoni et coll., 2002 ; Good et coll., 2002 ; Boxer et coll., 2003 ; Busatto et coll., 2003 ; Gee et coll., 2003 ; Karas et coll., 2003 ; Grossman et coll., 2004 ; Zahn et coll., 2005 ; Bozzali et coll., 2006 ; Kawachi et coll., 2006 ; Shiino et coll., 2006 ; Xie et coll., 2006) a montré que l'atrophie s'étendait au-delà du lobe temporal interne. Ainsi, à un stade léger à modéré de la maladie d'Alzheimer, l'atrophie touche non seulement la région hippocampique, mais aussi le cortex temporo-pariétal, le cortex cingulaire postérieur et le précuneus. L'atrophie d'autres structures est parfois observée, comme le cortex cingulaire antérieur, le cortex frontal, le thalamus, les noyaux caudés et les putamen ou l'insula. Chez des patients atteints de MCI, les études VBM ont également mis en évidence une atrophie du lobe temporal interne, ainsi que d'autres régions cérébrales (Chételat et coll., 2002 ; Karas et coll., 2004 ; Pennanen et coll., 2005). La perte de substance grise y est cependant moins marquée dans le MCI que dans la maladie d'Alzheimer (Chételat et coll., 2002 ; Karas et coll., 2004). Enfin, l'atrophie du cortex entorhinal (Hirata et coll., 2005), de l'hippocampe, des gyri parahippocampique et fusiforme, du cortex cingulaire postérieur et du précuneus (Chételat et coll., 2005a) est plus importante chez les converteurs que chez les non converteurs. Des études récentes ont tenté de spécifier la partie de la région hippocampique la plus atrophiée. Ainsi, l'atteinte du champ CA1 de l'hippocampe semble plus marquée chez les converteurs que chez les non converteurs (Apostolova et coll., 2006) et être le meilleur indice prédictif du déclin cognitif chez des patients à risque (Csernansky et coll., 2005).
), mais il s'agit d'une méthode subjective, qui demande de l'expertise et ne permet pas d'obtenir des données quantitatives du degré d'atrophie. La méthode des ROIs présente aussi des limites, dans la mesure où leur délimitation dépend de l'expertise de l'observateur. L'ensemble de ces méthodes ne permet pas d'étudier l'atrophie cérébrale de manière exhaustive et objective. Une méthode automatique et reproductible a donc été employée pour analyser l'ensemble du cerveau, voxel par voxel (Voxel Based Morphometry, VBM). Cette méthode, utilisée pour la première fois dans un groupe important de patients par Baron et coll. (2001), puis par de nombreux autres auteurs (Ohnishi et coll., 2001 ; Burton et coll., 2002 ; Frisoni et coll., 2002 ; Good et coll., 2002 ; Boxer et coll., 2003 ; Busatto et coll., 2003 ; Gee et coll., 2003 ; Karas et coll., 2003 ; Grossman et coll., 2004 ; Zahn et coll., 2005 ; Bozzali et coll., 2006 ; Kawachi et coll., 2006 ; Shiino et coll., 2006 ; Xie et coll., 2006) a montré que l'atrophie s'étendait au-delà du lobe temporal interne. Ainsi, à un stade léger à modéré de la maladie d'Alzheimer, l'atrophie touche non seulement la région hippocampique, mais aussi le cortex temporo-pariétal, le cortex cingulaire postérieur et le précuneus. L'atrophie d'autres structures est parfois observée, comme le cortex cingulaire antérieur, le cortex frontal, le thalamus, les noyaux caudés et les putamen ou l'insula. Chez des patients atteints de MCI, les études VBM ont également mis en évidence une atrophie du lobe temporal interne, ainsi que d'autres régions cérébrales (Chételat et coll., 2002 ; Karas et coll., 2004 ; Pennanen et coll., 2005). La perte de substance grise y est cependant moins marquée dans le MCI que dans la maladie d'Alzheimer (Chételat et coll., 2002 ; Karas et coll., 2004). Enfin, l'atrophie du cortex entorhinal (Hirata et coll., 2005), de l'hippocampe, des gyri parahippocampique et fusiforme, du cortex cingulaire postérieur et du précuneus (Chételat et coll., 2005a) est plus importante chez les converteurs que chez les non converteurs. Des études récentes ont tenté de spécifier la partie de la région hippocampique la plus atrophiée. Ainsi, l'atteinte du champ CA1 de l'hippocampe semble plus marquée chez les converteurs que chez les non converteurs (Apostolova et coll., 2006) et être le meilleur indice prédictif du déclin cognitif chez des patients à risque (Csernansky et coll., 2005).En résumé, l'atrophie évolue avec la sévérité de la maladie d'Alzheimer, du lobe temporal interne au néocortex, de façon superposable à la progression topographique des dégénérescences neurofibrillaires (DNF) (Duyckaerts et coll., 1998 ; Delacourte et coll., 1999). L'estimation de l'atrophie hippocampique varie entre 20 et 52 % dans la maladie d'Alzheimer et entre 11 et 23 % dans le MCI (Chételat et Baron, 2003). Son pouvoir discriminant est estimé à environ 70 à 80 % selon les études sur la maladie d'Alzheimer à un stade léger (Chételat et Baron, 2003). La combinaison de mesures améliore la discrimination entre les patients et les sujets sains. Ceci a été montré en combinant des mesures d'atrophie de l'hippocampe et de l'amygdale (Lehéricy et coll., 1994 ; Hampel et coll., 2002), ou du cortex entorhinal et du néocortex temporal (Killiany et coll., 2000).
; Delacourte et coll., 1999). L'estimation de l'atrophie hippocampique varie entre 20 et 52 % dans la maladie d'Alzheimer et entre 11 et 23 % dans le MCI (Chételat et Baron, 2003). Son pouvoir discriminant est estimé à environ 70 à 80 % selon les études sur la maladie d'Alzheimer à un stade léger (Chételat et Baron, 2003). La combinaison de mesures améliore la discrimination entre les patients et les sujets sains. Ceci a été montré en combinant des mesures d'atrophie de l'hippocampe et de l'amygdale (Lehéricy et coll., 1994 ; Hampel et coll., 2002), ou du cortex entorhinal et du néocortex temporal (Killiany et coll., 2000).Profil d'atteinte fonctionnelle
Des modifications fonctionnelles de certaines structures cérébrales ont été mises en évidence en utilisant la TEP au 18-fluorodésoxyglucose (TEP18FDG) ou la TEMP (tomographie d'émission monophotonique) au 99mTc-HMPAO (HéxaMéthyl Propylène Amine Oxime) ou au 99mTc-ECD (EthylCysteine Dimer) chez des patients au repos. Ces techniques mesurent des paramètres qui varient localement avec l'activité neuronale, et permettent donc l'étude de l'activité fonctionnelle du cerveau au niveau régional. La TEMP mesure le débit sanguin cérébral, tandis que la TEP-18FDG au repos, plus sensible, mesure la consommation cérébrale de glucose, paramètre qui reflète principalement l'activité synaptique. Cette technique permet ainsi de visualiser et de quantifier les modifications fonctionnelles locales dues soit à la perte synaptique locale, soit à un dysfonctionnement synaptique, invisible en imagerie structurale, induit par des désordres biochimiques ou reflétant l'effet à distance de lésions d'autres structures (par dysconnexion).
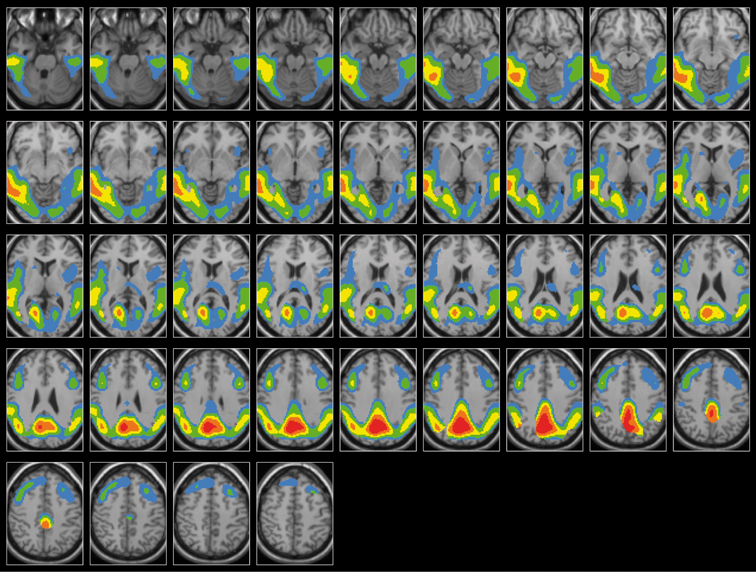
Le profil d'atteinte fonctionnelle dans la maladie d'Alzheimer est relativement constant à travers les études utilisant la méthode des ROIs ou la méthode SPM (Statistical Parametric Mapping) qui explore l'ensemble du cerveau de façon objective (Matsuda, 2001 ; Herholz, 2003 ; Nestor et coll., 2004 ; Mosconi, 2005 ; pour revues). Ainsi, sur le plan régional, l'hypométabolisme ou l'hypoperfusion du cortex temporo-pariétal et du gyrus cingulaire postérieur, constitue l'anomalie la plus précoce et la plus fréquente (Minoshima et coll., 1997 ; Kawachi et coll., 2006 ; voir Demetriades, 2002, pour revue). La baisse du métabolisme s'étend ensuite au cortex frontal (Desgranges et coll., 1998a ; Alexander et coll., 2002 ; Nestor et coll., 2003b ; Zahn et coll., 2005), mais le métabolisme du cortex primaire moteur et sensoriel, des noyaux gris centraux et du cervelet est relativement préservé (figure 7.1). Ce profil métabolique est en accord avec l'altération des fonctions cognitives et la préservation des fonctions sensorimotrices observées chez la majorité des patients.
; Herholz, 2003 ; Nestor et coll., 2004 ; Mosconi, 2005 ; pour revues). Ainsi, sur le plan régional, l'hypométabolisme ou l'hypoperfusion du cortex temporo-pariétal et du gyrus cingulaire postérieur, constitue l'anomalie la plus précoce et la plus fréquente (Minoshima et coll., 1997 ; Kawachi et coll., 2006 ; voir Demetriades, 2002, pour revue). La baisse du métabolisme s'étend ensuite au cortex frontal (Desgranges et coll., 1998a ; Alexander et coll., 2002 ; Nestor et coll., 2003b ; Zahn et coll., 2005), mais le métabolisme du cortex primaire moteur et sensoriel, des noyaux gris centraux et du cervelet est relativement préservé (figure 7.1). Ce profil métabolique est en accord avec l'altération des fonctions cognitives et la préservation des fonctions sensorimotrices observées chez la majorité des patients. | Figure 7.1 Hypométabolisme chez 40 patients atteints de maladie d'Alzheimer. (données de l'Unité Inserm E 0218 de Caen) |
Le dysfonctionnement du cortex cingulaire postérieur existe également chez des sujets sains porteurs de l'allèle ɛ4 du gène de l'apolipoprotéine E (APOE4), qui présentent un risque accru de développer la maladie d'Alzheimer, qu'ils soient âgés (Reiman et coll., 1996) ou jeunes (Reiman et coll., 2004). Les patients atteints de MCI, comparés à des sujets sains, présentent aussi un hypométabolisme (Chételat et coll., 2003b ; Nestor et coll., 2003b) ou une hypoperfusion (Hirao et coll., 2005) au niveau de cette région. De plus, son atteinte est plus importante chez les converteurs que chez les non converteurs (Huang et coll., 2002 ; Chételat et coll., 2003b ; Drzezga et coll., 2003 ; Hirao et coll., 2005), mais c'est le dysfonctionnement du cortex temporo-pariétal qui différencie le mieux ces deux populations de patients MCI, que ce soit en TEP-18FDG (Arnaiz et coll., 2001 ; Chételat et coll., 2003b ; Drzezga et coll., 2003 ; Mosconi et coll., 2004 ; Anchisi et coll., 2005) ou en TEMP (Huang et coll., 2003 ; Hirao et coll., 2005). De plus, l'hypométabolisme du cortex temporo-pariétal chez les converteurs a été montré à l'échelle individuelle (Nestor et coll., 2003a). Il permettrait de distinguer les converteurs des non converteurs avec une efficacité de 100 % (Chételat et coll., 2003b) et serait prédictif du déclin cognitif ultérieur (Chételat et coll., 2005b).
) ou jeunes (Reiman et coll., 2004). Les patients atteints de MCI, comparés à des sujets sains, présentent aussi un hypométabolisme (Chételat et coll., 2003b ; Nestor et coll., 2003b) ou une hypoperfusion (Hirao et coll., 2005) au niveau de cette région. De plus, son atteinte est plus importante chez les converteurs que chez les non converteurs (Huang et coll., 2002 ; Chételat et coll., 2003b ; Drzezga et coll., 2003 ; Hirao et coll., 2005), mais c'est le dysfonctionnement du cortex temporo-pariétal qui différencie le mieux ces deux populations de patients MCI, que ce soit en TEP-18FDG (Arnaiz et coll., 2001 ; Chételat et coll., 2003b ; Drzezga et coll., 2003 ; Mosconi et coll., 2004 ; Anchisi et coll., 2005) ou en TEMP (Huang et coll., 2003 ; Hirao et coll., 2005). De plus, l'hypométabolisme du cortex temporo-pariétal chez les converteurs a été montré à l'échelle individuelle (Nestor et coll., 2003a). Il permettrait de distinguer les converteurs des non converteurs avec une efficacité de 100 % (Chételat et coll., 2003b) et serait prédictif du déclin cognitif ultérieur (Chételat et coll., 2005b).Une asymétrie nette au niveau de l'atteinte du cortex temporo-pariétal est rapportée dans la plupart des études en TEP-18FDG, qui impliquent préférentiellement, voire exclusivement, l'hémisphère droit dans le MCI (Chételat et coll., 2003b ; Drzezga et coll., 2003; Mosconi et coll., 2004) ou dans la maladie d'Alzheimer à un stade léger (Nagahama et coll., 2003 ; Kawachi et coll., 2006 ; Chételat et coll., 2006, pour revue). Ce résultat suggère que lorsque l'hypométabolisme temporo-pariétal touche l'hémisphère droit, les patients sont à un stade débutant de la maladie d'Alzheimer, alors que lorsqu'il s'étend à l'hémisphère gauche, les patients sont à un stade plus sévère de la maladie. Plusieurs hypothèses peuvent être envisagées afin de rendre compte de ce phénomène. Premièrement, l'hémisphère droit pourrait être plus sensible aux atteintes fonctionnelles, il serait donc touché en premier. Les troubles cognitifs seraient alors encore mineurs, voire compensés par le recrutement d'autres régions cérébrales. L'hémisphère gauche ne serait altéré d'un point de vue fonctionnel que plus tard dans l'évolution de la maladie, induisant des troubles cognitifs plus notables. Deuxièmement, cette prédominance de l'atteinte droite pourrait refléter un biais lié aux tests utilisés pour diagnostiquer les patients. En effet, les évaluations sont principalement basées sur les capacités verbales, et seraient donc plus sensibles aux perturbations de l'hémisphère gauche, par opposition aux capacités visuo-spatiales, davantage sous-tendues par l'hémisphère droit. Par conséquent, une atteinte temporo-pariétale droite s'accompagnerait de déficits mineurs lors des évaluations, tandis qu'une atteinte équivalente à gauche induirait des troubles plus faciles à détecter par les tests, les patients n'étant alors pas diagnostiqués MCI, mais maladie d'Alzheimer probable.
; Drzezga et coll., 2003; Mosconi et coll., 2004) ou dans la maladie d'Alzheimer à un stade léger (Nagahama et coll., 2003 ; Kawachi et coll., 2006 ; Chételat et coll., 2006, pour revue). Ce résultat suggère que lorsque l'hypométabolisme temporo-pariétal touche l'hémisphère droit, les patients sont à un stade débutant de la maladie d'Alzheimer, alors que lorsqu'il s'étend à l'hémisphère gauche, les patients sont à un stade plus sévère de la maladie. Plusieurs hypothèses peuvent être envisagées afin de rendre compte de ce phénomène. Premièrement, l'hémisphère droit pourrait être plus sensible aux atteintes fonctionnelles, il serait donc touché en premier. Les troubles cognitifs seraient alors encore mineurs, voire compensés par le recrutement d'autres régions cérébrales. L'hémisphère gauche ne serait altéré d'un point de vue fonctionnel que plus tard dans l'évolution de la maladie, induisant des troubles cognitifs plus notables. Deuxièmement, cette prédominance de l'atteinte droite pourrait refléter un biais lié aux tests utilisés pour diagnostiquer les patients. En effet, les évaluations sont principalement basées sur les capacités verbales, et seraient donc plus sensibles aux perturbations de l'hémisphère gauche, par opposition aux capacités visuo-spatiales, davantage sous-tendues par l'hémisphère droit. Par conséquent, une atteinte temporo-pariétale droite s'accompagnerait de déficits mineurs lors des évaluations, tandis qu'une atteinte équivalente à gauche induirait des troubles plus faciles à détecter par les tests, les patients n'étant alors pas diagnostiqués MCI, mais maladie d'Alzheimer probable.Par ailleurs, alors que l'atteinte structurale de l'hippocampe est précoce et marquée, son atteinte fonctionnelle est loin d'être constamment observée, ce qui constitue un véritable paradoxe, et suscite des débats dans la littérature (Ishii et coll., 1998 ; Desgranges et coll., 2004 ; Chételat et coll., 2006, pour revues). Ainsi, elle n'a pas été mise en évidence dans une étude portant sur près de 400 patients (Herholz et coll., 2002). De façon paradoxale, une hypoperfusion de cette région a été mise en évidence en TEMP (Desgranges et coll., 2004, pour revue), mais il pourrait s'agir d'un artefact dû à l'« effet de volume partiel » c'est-à-dire à la sous-estimation de l'activité synaptique pour certaines régions de petite taille ou atrophiques due aux faibles résolutions spatiales de ces techniques d'imagerie (et donc plus importantes avec la TEMP), comme le suggère l'étude de Matsuda et coll. (2002). Des méthodes permettent actuellement de corriger cet effet, en TEP, comme en TEMP. Ainsi, Ibanez et coll. (1998) et Bokde et coll. (2001) ont montré dans la maladie d'Alzheimer une persistance de l'hypométabolisme temporo-pariétal et cingulaire postérieur après avoir corrigé les valeurs de consommation de glucose de l'effet de volume partiel. À l'inverse, Matsuda et coll. (2002) ont montré que l'hypodébit hippocampique observé avant correction des effets de volume partiel disparaissait après cette correction. D'autres questions méthodologiques ont été soulevées, par exemple la méthode des régions d'intérêt détecte mieux l'hypométabolisme de l'hippocampe que la méthode SPM (Nestor et coll., 2003b ; Mosconi et coll., 2005 ; Edison et coll., 2007). L'effet de différents facteurs méthodologiques a été testé dans une étude récente qui montre un hypométabolisme hippocampique grâce à l'approche voxel par voxel (Mevel et coll., sous presse). Quoi qu'il en soit, lorsqu'un dysfonctionnement de l'hippocampe est détecté, il est toujours moins important que dans les régions corticales, il n'est jamais le plus précoce et il est plus facile à mettre en évidence chez les patients atteints de démence modérée que légère (Kogure et coll., 2000 ; Kitayama et coll., 2001 ; Mosconi, 2005).
; Desgranges et coll., 2004 ; Chételat et coll., 2006, pour revues). Ainsi, elle n'a pas été mise en évidence dans une étude portant sur près de 400 patients (Herholz et coll., 2002). De façon paradoxale, une hypoperfusion de cette région a été mise en évidence en TEMP (Desgranges et coll., 2004, pour revue), mais il pourrait s'agir d'un artefact dû à l'« effet de volume partiel » c'est-à-dire à la sous-estimation de l'activité synaptique pour certaines régions de petite taille ou atrophiques due aux faibles résolutions spatiales de ces techniques d'imagerie (et donc plus importantes avec la TEMP), comme le suggère l'étude de Matsuda et coll. (2002). Des méthodes permettent actuellement de corriger cet effet, en TEP, comme en TEMP. Ainsi, Ibanez et coll. (1998) et Bokde et coll. (2001) ont montré dans la maladie d'Alzheimer une persistance de l'hypométabolisme temporo-pariétal et cingulaire postérieur après avoir corrigé les valeurs de consommation de glucose de l'effet de volume partiel. À l'inverse, Matsuda et coll. (2002) ont montré que l'hypodébit hippocampique observé avant correction des effets de volume partiel disparaissait après cette correction. D'autres questions méthodologiques ont été soulevées, par exemple la méthode des régions d'intérêt détecte mieux l'hypométabolisme de l'hippocampe que la méthode SPM (Nestor et coll., 2003b ; Mosconi et coll., 2005 ; Edison et coll., 2007). L'effet de différents facteurs méthodologiques a été testé dans une étude récente qui montre un hypométabolisme hippocampique grâce à l'approche voxel par voxel (Mevel et coll., sous presse). Quoi qu'il en soit, lorsqu'un dysfonctionnement de l'hippocampe est détecté, il est toujours moins important que dans les régions corticales, il n'est jamais le plus précoce et il est plus facile à mettre en évidence chez les patients atteints de démence modérée que légère (Kogure et coll., 2000 ; Kitayama et coll., 2001 ; Mosconi, 2005).L'hypométabolisme de l'hippocampe serait plus fréquent chez les patients porteurs du gène de l'APOE4 ayant débuté leur maladie avant l'âge de 65 ans (Hirono et coll., 2002). Plusieurs autres études se sont intéressées à l'influence de ce facteur sur les perturbations du métabolisme cérébral, et convergent vers des atteintes plus marquées chez les patients « à début précoce ». Selon les études, l'altération concerne le cortex frontal (Kim et coll., 2005 ; Kalpouzos et coll., 2005), le cortex pariétal (Sakamoto et coll., 2002 ; Kim et coll., 2005 ; Kalpouzos et coll., 2005), les noyaux gris (Kim et coll., 2005), ou le cortex cingulaire postérieur (Salmon et coll., 2000 ; Sakamoto et coll., 2002). Ces différences existent même lorsque les groupes sont appariés en termes de sévérité de la maladie, ce qui suggère l'existence de « réserves cognitives » qui leur permettraient de compenser en partie leurs déficits. L'altération plus importante chez les sujets jeunes est également en accord avec l'hypothèse d'un déclin plus rapide chez ces patients (Kim et coll., 2005).
). Plusieurs autres études se sont intéressées à l'influence de ce facteur sur les perturbations du métabolisme cérébral, et convergent vers des atteintes plus marquées chez les patients « à début précoce ». Selon les études, l'altération concerne le cortex frontal (Kim et coll., 2005 ; Kalpouzos et coll., 2005), le cortex pariétal (Sakamoto et coll., 2002 ; Kim et coll., 2005 ; Kalpouzos et coll., 2005), les noyaux gris (Kim et coll., 2005), ou le cortex cingulaire postérieur (Salmon et coll., 2000 ; Sakamoto et coll., 2002). Ces différences existent même lorsque les groupes sont appariés en termes de sévérité de la maladie, ce qui suggère l'existence de « réserves cognitives » qui leur permettraient de compenser en partie leurs déficits. L'altération plus importante chez les sujets jeunes est également en accord avec l'hypothèse d'un déclin plus rapide chez ces patients (Kim et coll., 2005).En résumé, le profil d'atteinte fonctionnelle est dominé par l'hypométabolisme du cortex cingulaire postérieur et temporo-pariétal. Des études récentes ont souligné la similitude entre ce profil d'altération fonctionnelle et le « réseau à l'état de base » (REB) mis en évidence par l'étude des désactivations observées lors de tâches cognitives chez le sujet sain (Buckner et coll., 2005). Ce REB serait constitué d'un ensemble de régions, plus activées au repos que lors de sollicitations cognitives, et sous-tendrait le fonctionnement « par défaut » du cerveau (Gusnard et coll., 2001 ; Greicius et coll., 2004 ; Fouquet et coll., sous presse, pour revue).
). Ce REB serait constitué d'un ensemble de régions, plus activées au repos que lors de sollicitations cognitives, et sous-tendrait le fonctionnement « par défaut » du cerveau (Gusnard et coll., 2001 ; Greicius et coll., 2004 ; Fouquet et coll., sous presse, pour revue).La sensibilité de la TEP aux altérations cérébrales de la maladie d'Alzheimer est estimée à plus de 90 % et sa spécificité à plus de 70 % (Silverman et coll., 2001), voire davantage (Herholz, 2003 ; Devous, 2002 ; Small, 2004, pour revues). La supériorité de la TEP sur les performances neuropsychologiques, mais également sur les mesures d'atrophie en IRM ou de perfusion en TEMP est bien établie (Drzezga et coll., 2003 ; Zamrini et coll., 2004 ; Chételat et coll., 2005b ; Hirao et coll., 2005 ; Kawachi et coll., 2006).
), voire davantage (Herholz, 2003 ; Devous, 2002 ; Small, 2004, pour revues). La supériorité de la TEP sur les performances neuropsychologiques, mais également sur les mesures d'atrophie en IRM ou de perfusion en TEMP est bien établie (Drzezga et coll., 2003 ; Zamrini et coll., 2004 ; Chételat et coll., 2005b ; Hirao et coll., 2005 ; Kawachi et coll., 2006).Discordance entre les profils
À un stade débutant de la maladie, le dysfonctionnement des régions cingulaire postérieure et temporo-pariétale contraste donc avec l'atrophie de la région hippocampique. Les hypothèses avancées afin d'expliquer cette discordance suggèrent d'une part un phénomène de dysconnexion et, d'autre part des mécanismes de compensation fonctionnelle (Chételat et coll., 2006). Plus précisément, les DNF et l'atrophie au niveau de la région hippocampique pourraient induire l'altération fonctionnelle des régions qui lui sont connectées, comme les cortex cingulaire postérieur et temporo-pariétal, par dysconnexion. Cette hypothèse, déjà avancée par Minoshima et coll. (1997), est étayée par les résultats d'une étude chez le babouin attestant de l'hypométabolisme des cortex temporo-pariétal et cingulaire postérieur après une lésion par neurotoxines au niveau du cortex entorhinal (Meguro et coll., 1999), et d'une étude chez l'homme montrant une corrélation entre l'hypométabolisme de la région temporo-pariétale et l'atrophie de l'hippocampe (Meguro et coll., 2001). La mise en évidence de corrélations significatives entre la densité de substance grise au niveau de l'hippocampe et l'activité du cortex cingulaire postérieur lors d'une tâche de mémoire (Garrido et coll., 2002 ; Rémy et coll., 2005) va également dans ce sens. Quant à la relative préservation fonctionnelle de la région hippocampique, elle pourrait résulter d'une compensation fonctionnelle liée à la plasticité élevée de cette région. Une augmentation de la neurogenèse (Jin et coll., 2004) ainsi qu'un rétro-contrôle cholinergique positif (DeKosky et coll., 2002) hippocampiques ont effectivement été mis en évidence dans la maladie d'Alzheimer. Cette idée trouve également un support dans les travaux récents de Dickerson et coll. (2005), réalisés en IRMf chez des patients atteints de MCI qui présentent des activations hippocampiques plus importantes que chez les sujets sains (voir infra). Les auteurs interprètent ces activations accrues comme une réponse compensatoire à la survenue de la maladie. La nature exacte de ces phénomènes reste à déterminer : modifications du couplage neuro-vasculaire liées à la maladie ou véritable compensation fonctionnelle reposant sur l'expansion des synapses restantes et le recrutement (voire la formation) de nouveaux neurones. Ces arguments indirects permettent d'appuyer les hypothèses proposées pour interpréter la discordance entre les atteintes structurales et fonctionnelles, mais elle reste encore mal comprise.
). Plus précisément, les DNF et l'atrophie au niveau de la région hippocampique pourraient induire l'altération fonctionnelle des régions qui lui sont connectées, comme les cortex cingulaire postérieur et temporo-pariétal, par dysconnexion. Cette hypothèse, déjà avancée par Minoshima et coll. (1997), est étayée par les résultats d'une étude chez le babouin attestant de l'hypométabolisme des cortex temporo-pariétal et cingulaire postérieur après une lésion par neurotoxines au niveau du cortex entorhinal (Meguro et coll., 1999), et d'une étude chez l'homme montrant une corrélation entre l'hypométabolisme de la région temporo-pariétale et l'atrophie de l'hippocampe (Meguro et coll., 2001). La mise en évidence de corrélations significatives entre la densité de substance grise au niveau de l'hippocampe et l'activité du cortex cingulaire postérieur lors d'une tâche de mémoire (Garrido et coll., 2002 ; Rémy et coll., 2005) va également dans ce sens. Quant à la relative préservation fonctionnelle de la région hippocampique, elle pourrait résulter d'une compensation fonctionnelle liée à la plasticité élevée de cette région. Une augmentation de la neurogenèse (Jin et coll., 2004) ainsi qu'un rétro-contrôle cholinergique positif (DeKosky et coll., 2002) hippocampiques ont effectivement été mis en évidence dans la maladie d'Alzheimer. Cette idée trouve également un support dans les travaux récents de Dickerson et coll. (2005), réalisés en IRMf chez des patients atteints de MCI qui présentent des activations hippocampiques plus importantes que chez les sujets sains (voir infra). Les auteurs interprètent ces activations accrues comme une réponse compensatoire à la survenue de la maladie. La nature exacte de ces phénomènes reste à déterminer : modifications du couplage neuro-vasculaire liées à la maladie ou véritable compensation fonctionnelle reposant sur l'expansion des synapses restantes et le recrutement (voire la formation) de nouveaux neurones. Ces arguments indirects permettent d'appuyer les hypothèses proposées pour interpréter la discordance entre les atteintes structurales et fonctionnelles, mais elle reste encore mal comprise.Liens avec les troubles cognitifs
Deux types d'approche permettent d'étudier le rôle des altérations cérébrales dans les troubles cognitifs.
Corrélations cognitivo-morphologiques et cognitivo-métaboliques
Cette approche qui consiste à établir des liens entre des altérations cérébrales et des troubles neuropsychologiques, a pour objectif d'identifier les régions cérébrales dont le dysfonctionnement est responsable de troubles cognitifs spécifiques (Desgranges et coll., 2004, pour revue). Des liens entre l'atrophie de l'hippocampe et des déficits de mémoire épisodique antérograde (Deweer et coll., 1995 ; Pantel et coll., 2004) et rétrograde (Gilboa et coll., 2005) ont ainsi été mis en évidence chez des patients atteints de maladie d'Alzheimer, à l'aide de la méthode des ROIs. Chez des patients atteints de MCI, l'analyse voxel par voxel a également montré des corrélations significatives entre la densité de substance grise de l'hippocampe et des scores de mémoire épisodique (Chételat et coll., 2003a). Grossman et coll. (2004) ont aussi montré des liens entre des performances de dénomination d'images et la densité de substance grise dans le lobe temporal antérieur gauche.
, pour revue). Des liens entre l'atrophie de l'hippocampe et des déficits de mémoire épisodique antérograde (Deweer et coll., 1995 ; Pantel et coll., 2004) et rétrograde (Gilboa et coll., 2005) ont ainsi été mis en évidence chez des patients atteints de maladie d'Alzheimer, à l'aide de la méthode des ROIs. Chez des patients atteints de MCI, l'analyse voxel par voxel a également montré des corrélations significatives entre la densité de substance grise de l'hippocampe et des scores de mémoire épisodique (Chételat et coll., 2003a). Grossman et coll. (2004) ont aussi montré des liens entre des performances de dénomination d'images et la densité de substance grise dans le lobe temporal antérieur gauche.L'approche corrélative a surtout été utilisée avec des mesures métaboliques, à l'aide de la méthode ROIs (Perani et coll., 1993) et de la méthode SPM (Desgranges et coll., 1998b et 2002 ; Chételat et coll., 2003a ; Lekeu et coll., 2003 ; Eustache et coll., 2001 et 2004 ; Teipel et coll., 2006 ; Rauchs et coll., sous presse). Ces travaux ont montré que le dysfonctionnement de différentes régions cérébrales était responsable des troubles de différents systèmes de mémoire (Desgranges et coll., 1998). Les troubles précoces de la mémoire épisodique sont sous-tendus par le dysfonctionnement de la région hippocampique (Eustache et coll., 2001 ; 2004 ; Lekeu et coll., 2003), tandis que les troubles de la mémoire sémantique sont liés au dysfonctionnement du cortex temporal gauche (Desgranges et coll., 1998 ; Hirono et coll., 2001). Dans la maladie d'Alzheimer à un stade modéré, cette approche a aussi permis de souligner l'existence de mécanismes compensatoires mis en jeu dans une tâche de mémoire épisodique. Ces mécanismes seraient sous-tendus par des régions néocorticales temporales, normalement dévolues à la mémoire sémantique (Desgranges et coll., 2002). Chez des patients atteints de MCI, une étude a montré que les troubles de l'encodage et de la récupération semblent dépendre de deux structures cérébrales différentes, respectivement l'hippocampe et le gyrus cingulaire postérieur (Chételat et coll., 2003a) (figure 7.2), deux structures-clés dans le fonctionnement normal de la mémoire épisodique (voir Cabeza et Nyberg, 2000, pour revue). D'autres déficits cognitifs ont été mis en relation avec des troubles de la perfusion ou du métabolisme, notamment la dyscalculie et le métabolisme du cortex temporal et pariétal gauches (Hirono et coll., 1998).
) et de la méthode SPM (Desgranges et coll., 1998b et 2002 ; Chételat et coll., 2003a ; Lekeu et coll., 2003 ; Eustache et coll., 2001 et 2004 ; Teipel et coll., 2006 ; Rauchs et coll., sous presse). Ces travaux ont montré que le dysfonctionnement de différentes régions cérébrales était responsable des troubles de différents systèmes de mémoire (Desgranges et coll., 1998). Les troubles précoces de la mémoire épisodique sont sous-tendus par le dysfonctionnement de la région hippocampique (Eustache et coll., 2001 ; 2004 ; Lekeu et coll., 2003), tandis que les troubles de la mémoire sémantique sont liés au dysfonctionnement du cortex temporal gauche (Desgranges et coll., 1998 ; Hirono et coll., 2001). Dans la maladie d'Alzheimer à un stade modéré, cette approche a aussi permis de souligner l'existence de mécanismes compensatoires mis en jeu dans une tâche de mémoire épisodique. Ces mécanismes seraient sous-tendus par des régions néocorticales temporales, normalement dévolues à la mémoire sémantique (Desgranges et coll., 2002). Chez des patients atteints de MCI, une étude a montré que les troubles de l'encodage et de la récupération semblent dépendre de deux structures cérébrales différentes, respectivement l'hippocampe et le gyrus cingulaire postérieur (Chételat et coll., 2003a) (figure 7.2), deux structures-clés dans le fonctionnement normal de la mémoire épisodique (voir Cabeza et Nyberg, 2000, pour revue). D'autres déficits cognitifs ont été mis en relation avec des troubles de la perfusion ou du métabolisme, notamment la dyscalculie et le métabolisme du cortex temporal et pariétal gauches (Hirono et coll., 1998).

Études en activation
La plupart des études d'activation en TEP ou en IRMf effectuées dans le cadre de la maladie d'Alzheimer s'intéressent à la mémoire épisodique, mesurant les modifications de l'activité cérébrale pendant l'encodage, ou pendant la récupération d'informations. En dépit de résultats parfois discordants, une première observation consensuelle émerge : la diminution des activations hippocampiques, tant lors de l'encodage que lors de la récupération (Desgranges et Eustache, 2005 ; Fouquet et coll., / sous presse, pour revues) chez les patients atteints de maladie d'Alzheimer par rapport aux témoins (Bäckman et coll., 1999 ; Rombouts et coll., 2000 ; Kato et coll., 2001 ; Garrido et coll., 2002 ; Grön et coll., 2002 ; Machulda et coll., 2003 ; Sperling et coll., 2003 ; Dickerson et coll., 2005 ; Golby et coll., 2005 ; Pariente et coll., 2005 ; Rémy et coll., 2005 ; Starr et coll., 2005 ; Celone et coll., 2006 ; Hämäläinen et coll., sous presse). Cette hypoactivation semble réellement témoigner d'une altération fonctionnelle puisqu'elle est retrouvée même lorsque les différences de volume hippocampique sont contrôlées entre les groupes (Dickerson et coll., 2005).
; Fouquet et coll., / sous presse, pour revues) chez les patients atteints de maladie d'Alzheimer par rapport aux témoins (Bäckman et coll., 1999 ; Rombouts et coll., 2000 ; Kato et coll., 2001 ; Garrido et coll., 2002 ; Grön et coll., 2002 ; Machulda et coll., 2003 ; Sperling et coll., 2003 ; Dickerson et coll., 2005 ; Golby et coll., 2005 ; Pariente et coll., 2005 ; Rémy et coll., 2005 ; Starr et coll., 2005 ; Celone et coll., 2006 ; Hämäläinen et coll., sous presse). Cette hypoactivation semble réellement témoigner d'une altération fonctionnelle puisqu'elle est retrouvée même lorsque les différences de volume hippocampique sont contrôlées entre les groupes (Dickerson et coll., 2005).Chez des patients à risque accru de développer la maladie d'Alzheimer, c'est-à-dire des sujets asymptomatiques porteurs d'au moins un allèle ɛ4 du gène de l'apolipoprotéine E (Bookheimer et coll., 2000 ; Bondi et coll., 2005 ; Han et coll., 2007), des sujets porteurs de cet allèle et présentant une histoire familiale de maladie d'Alzheimer sporadique (Fleisher et coll., 2005 ; Bassett et coll., 2006 ; Johnson et coll., 2006b) ou des patients atteints de MCI (Dickerson et coll., 2004 et 2005 ; Celone et coll., 2006 ; Hämäläinen et coll., sous presse ; Kircher et coll., sous presse), l'activité de la région hippocampique semble au contraire augmentée, par rapport à des sujets témoins à l'occasion de tâches de mémoire épisodique. De plus, Bookheimer et coll. (2000) et Dickerson et coll. (2004) ont montré le caractère prédictif de l'hyperactivité hippocampique sur le déclin cognitif ultérieur des sujets.
; Bondi et coll., 2005 ; Han et coll., 2007), des sujets porteurs de cet allèle et présentant une histoire familiale de maladie d'Alzheimer sporadique (Fleisher et coll., 2005 ; Bassett et coll., 2006 ; Johnson et coll., 2006b) ou des patients atteints de MCI (Dickerson et coll., 2004 et 2005 ; Celone et coll., 2006 ; Hämäläinen et coll., sous presse ; Kircher et coll., sous presse), l'activité de la région hippocampique semble au contraire augmentée, par rapport à des sujets témoins à l'occasion de tâches de mémoire épisodique. De plus, Bookheimer et coll. (2000) et Dickerson et coll. (2004) ont montré le caractère prédictif de l'hyperactivité hippocampique sur le déclin cognitif ultérieur des sujets.Cette hyperactivation hippocampique aux stades précoces de la maladie demeure néanmoins controversée puisque plusieurs études ont montré, lors de tâches de mémoire épisodique, des activations hippocampiques équivalentes entre les groupes (Sandström et coll., 2006 ; Heun et coll., 2007) ou au contraire inférieures chez les sujets à risque (Machulda et coll., 2003 ; Grön et coll., 2004 ; Johnson et coll., 2006a ; Lind et coll., 2006 ; Trivedi et coll., 2006). L'hétérogénéité des résultats s'explique en partie par la diversité des méthodologies employées ainsi que par des différences de stade de la maladie. En effet, l'hyperactivation hippocampique compensatoire pourrait être transitoire et se manifester uniquement à un stade précoce. Cette activité pourrait par la suite chuter en dessous de celle des témoins avec l'évolution de la maladie (Prvulovic et coll., 2005). Deux travaux récents semblent confirmer cette hypothèse. Celone et coll. (2006) ont montré que des patients atteints de MCI dont les performances cognitives sont plus altérées présentent une hypoactivation hippocampique du même niveau que celle des patients atteints de la maladie d'Alzheimer alors que des MCI dont les performances cognitives sont moins altérées présentent au contraire une hyperactivation hippocampique. Dans une étude de cas portant sur des sujets d'une famille porteuse d'une mutation génétique d'une forme familiale de la maladie d'Alzheimer, Mondadori et coll. (2006) rapportent quant à eux une hyperactivation hippocampique chez un sujet jeune dont les performances cognitives sont normales et une hypoactivation de l'hippocampe chez un sujet de 45 ans qui remplissait les critères diagnostiques de MCI amnésique.
; Heun et coll., 2007) ou au contraire inférieures chez les sujets à risque (Machulda et coll., 2003 ; Grön et coll., 2004 ; Johnson et coll., 2006a ; Lind et coll., 2006 ; Trivedi et coll., 2006). L'hétérogénéité des résultats s'explique en partie par la diversité des méthodologies employées ainsi que par des différences de stade de la maladie. En effet, l'hyperactivation hippocampique compensatoire pourrait être transitoire et se manifester uniquement à un stade précoce. Cette activité pourrait par la suite chuter en dessous de celle des témoins avec l'évolution de la maladie (Prvulovic et coll., 2005). Deux travaux récents semblent confirmer cette hypothèse. Celone et coll. (2006) ont montré que des patients atteints de MCI dont les performances cognitives sont plus altérées présentent une hypoactivation hippocampique du même niveau que celle des patients atteints de la maladie d'Alzheimer alors que des MCI dont les performances cognitives sont moins altérées présentent au contraire une hyperactivation hippocampique. Dans une étude de cas portant sur des sujets d'une famille porteuse d'une mutation génétique d'une forme familiale de la maladie d'Alzheimer, Mondadori et coll. (2006) rapportent quant à eux une hyperactivation hippocampique chez un sujet jeune dont les performances cognitives sont normales et une hypoactivation de l'hippocampe chez un sujet de 45 ans qui remplissait les critères diagnostiques de MCI amnésique.Un deuxième résultat émerge des études d'activation dans la maladie d'Alzheimer : l'augmentation des activations, principalement situées dans le cortex frontal et supposées sous-tendre les mécanismes compensatoires. Des augmentations d'activations frontales sont effectivement très souvent rapportées (Grady et coll., 1993 ; Becker et coll., 1996 ; Woodard et coll., 1998 ; Bäckman et coll., 1999 ; Garrido et coll., 2002 ; Grön et coll., 2002 ; Grossman et coll., 2003 ; Sperling et coll., 2003 ; Hao et coll., 2005 ; Pariente et coll., 2005 ; Rémy et coll., 2005 ; Starr et coll., 2005 ; Xie et coll., 2005a ; Yetkin et coll., 2006). Elles pourraient s'expliquer par l'atteinte plus tardive de cette région cérébrale, et sont souvent considérées comme le témoin de la mise en œ uvre plus importante des ressources attentionnelles et/ou des fonctions exécutives par les patients pour effectuer la même tâche que les sujets âgés sains. L'hyperactivation de la région frontale a également été mise en évidence chez des patients MCI, lors de tâches de mémoire épisodique (Heun et coll., 2007), de mémoire de travail (Wishart et coll., 2006) ou de tâche attentionnelle (Rosano et coll., 2005).
; Becker et coll., 1996 ; Woodard et coll., 1998 ; Bäckman et coll., 1999 ; Garrido et coll., 2002 ; Grön et coll., 2002 ; Grossman et coll., 2003 ; Sperling et coll., 2003 ; Hao et coll., 2005 ; Pariente et coll., 2005 ; Rémy et coll., 2005 ; Starr et coll., 2005 ; Xie et coll., 2005a ; Yetkin et coll., 2006). Elles pourraient s'expliquer par l'atteinte plus tardive de cette région cérébrale, et sont souvent considérées comme le témoin de la mise en œ uvre plus importante des ressources attentionnelles et/ou des fonctions exécutives par les patients pour effectuer la même tâche que les sujets âgés sains. L'hyperactivation de la région frontale a également été mise en évidence chez des patients MCI, lors de tâches de mémoire épisodique (Heun et coll., 2007), de mémoire de travail (Wishart et coll., 2006) ou de tâche attentionnelle (Rosano et coll., 2005).Cependant, les différences observées entre patients Alzheimer et sujets sains sont parfois de faible amplitude, notamment en cas de contrôle de la différence de complexité des tâches (Gould et coll., 2005), voire inexistantes ou en faveur des sujets sains (Saykin et coll., 1999 ; Stern et coll., 2000 ; Kato et coll., 2001 ; Schröder et coll., 2001 ; Elgh et coll., 2003 ; Golby et coll., 2005). Dans certaines études, l'hyperactivation frontale n'est observée qu'à un stade prédémentiel et le sens de la différence s'inverse au stade de démence (Celone et coll., 2006 ; Hämäläinen et coll., sous presse). Ces résultats soulignent les difficultés et limites des simples comparaisons de groupe pour mettre en évidence les phénomènes de compensation cérébrale. De plus, dans la plupart des études, l'efficacité des « mécanismes compensatoires » n'était pas établie et semblait pour le moins insuffisante, puisque le niveau moyen de performance des patients était inférieur à celui des témoins. Quelques études ont pourtant conforté cette hypothèse en montrant d'une part une corrélation négative entre le volume hippocampique et l'activité corticale au sein du lobe frontal (Johnson et coll., 2000 ; Garrido et coll., 2002 ; Rémy et coll., 2005) et d'autre part des corrélations positives entre les activations frontales et les performances mnésiques (Rémy et coll., 2005, voir aussi Grady et coll., 2003 ; Golby et coll., 2005).
), voire inexistantes ou en faveur des sujets sains (Saykin et coll., 1999 ; Stern et coll., 2000 ; Kato et coll., 2001 ; Schröder et coll., 2001 ; Elgh et coll., 2003 ; Golby et coll., 2005). Dans certaines études, l'hyperactivation frontale n'est observée qu'à un stade prédémentiel et le sens de la différence s'inverse au stade de démence (Celone et coll., 2006 ; Hämäläinen et coll., sous presse). Ces résultats soulignent les difficultés et limites des simples comparaisons de groupe pour mettre en évidence les phénomènes de compensation cérébrale. De plus, dans la plupart des études, l'efficacité des « mécanismes compensatoires » n'était pas établie et semblait pour le moins insuffisante, puisque le niveau moyen de performance des patients était inférieur à celui des témoins. Quelques études ont pourtant conforté cette hypothèse en montrant d'une part une corrélation négative entre le volume hippocampique et l'activité corticale au sein du lobe frontal (Johnson et coll., 2000 ; Garrido et coll., 2002 ; Rémy et coll., 2005) et d'autre part des corrélations positives entre les activations frontales et les performances mnésiques (Rémy et coll., 2005, voir aussi Grady et coll., 2003 ; Golby et coll., 2005).En résumé, au stade précoce de la maladie, la région hippocampique semble être le siège de modifications fonctionnelles compensatoires, tandis qu'à un stade plus avancé, les mécanismes compensatoires seraient pris en charge par des régions néocorticales, principalement frontales, parfois pariétales (Pariente et coll., 2005 ; Wishart et coll., 2006). Les résultats concernant les autres régions cérébrales sont plus divergents, aucune altération fonctionnelle cérébrale n'étant retrouvée de manière consensuelle. Quelques études rapportent cependant des activations plus faibles chez les patients au niveau du cortex cingulaire postérieur lors de tâches d'encodage (Gould et coll., 2005 ; Rémy et coll., 2005) ou de récupération (Johnson et coll., 2006a ; Ries et coll., 2006) en mémoire. Une hyperactivation de cette région a également été observée chez des sujets porteurs du gène de l'APOE4 (Han et coll., 2007) ou des patients atteints de maladie d'Alzheimer (Sperling et coll., 2003). De plus, le défaut d'activation de cette région apparaît dépendant de la tâche, puisqu'elle s'active normalement lors d'une tâche d'auto-évaluation à partir d'adjectifs fournis et non lors de la récupération mnésique des adjectifs (Ries et coll., 2006).
; Wishart et coll., 2006). Les résultats concernant les autres régions cérébrales sont plus divergents, aucune altération fonctionnelle cérébrale n'étant retrouvée de manière consensuelle. Quelques études rapportent cependant des activations plus faibles chez les patients au niveau du cortex cingulaire postérieur lors de tâches d'encodage (Gould et coll., 2005 ; Rémy et coll., 2005) ou de récupération (Johnson et coll., 2006a ; Ries et coll., 2006) en mémoire. Une hyperactivation de cette région a également été observée chez des sujets porteurs du gène de l'APOE4 (Han et coll., 2007) ou des patients atteints de maladie d'Alzheimer (Sperling et coll., 2003). De plus, le défaut d'activation de cette région apparaît dépendant de la tâche, puisqu'elle s'active normalement lors d'une tâche d'auto-évaluation à partir d'adjectifs fournis et non lors de la récupération mnésique des adjectifs (Ries et coll., 2006).Enfin, il faut garder à l'esprit que l'activité neuronale mesurée par l'imagerie cérébrale fonctionnelle en activation reste une mesure indirecte. En effet, le signal enregistré n'est que le reflet des modifications vasculaires à proximité des neurones activés. Ainsi, D'Esposito et coll. (2003) rappellent qu'il faut rester prudent car les modifications observées en imagerie fonctionnelle hémodynamique pourraient également témoigner d'altérations du couplage neuro-vasculaire liées à la maladie d'Alzheimer. De fait, les dépôts amyloïdes extraneuronaux caractéristiques de la maladie sont aussi présents au niveau vasculaire et pourraient ainsi interférer avec le couplage neuro-vasculaire (Iadecola, 2004). L'imagerie cérébrale en activation est donc un outil précieux dans l'étude de la maladie d'Alzheimer puisqu'elle permet de mieux comprendre le rôle des altérations cérébrales dans les troubles cognitifs et de mettre en évidence les compensations fonctionnelles mises en place par les patients. Toutefois, les observations demeurent hétérogènes et de nombreux aspects restent à préciser et à comprendre.
) rappellent qu'il faut rester prudent car les modifications observées en imagerie fonctionnelle hémodynamique pourraient également témoigner d'altérations du couplage neuro-vasculaire liées à la maladie d'Alzheimer. De fait, les dépôts amyloïdes extraneuronaux caractéristiques de la maladie sont aussi présents au niveau vasculaire et pourraient ainsi interférer avec le couplage neuro-vasculaire (Iadecola, 2004). L'imagerie cérébrale en activation est donc un outil précieux dans l'étude de la maladie d'Alzheimer puisqu'elle permet de mieux comprendre le rôle des altérations cérébrales dans les troubles cognitifs et de mettre en évidence les compensations fonctionnelles mises en place par les patients. Toutefois, les observations demeurent hétérogènes et de nombreux aspects restent à préciser et à comprendre.Études en IRM du tenseur de diffusion
Le développement de l'IRM du tenseur de diffusion (DTI), depuis le milieu des années 1980, offre la possibilité d'étudier l'intégrité structurale des fibres de la substance blanche et permet ainsi d'étudier la connectivité fonctionnelle de différentes régions cérébrales. La DTI est une technique qui repose sur les propriétés intrinsèques des mouvements de l'eau (Le Bihan et coll., 2001). Dans un tissu, la diffusion des molécules d'eau ne se fait pas de façon aléatoire mais elle est contrainte par différents facteurs environnants (les membranes cellulaires, les fibres axonales et diverses macromolécules). Leur diffusion dans la substance blanche, riche en fibres axonales, correspond donc à une diffusion anisotropique qui peut être caractérisée en DTI, grâce à deux indices de cette diffusion anisotropique : la diffusivité qui évalue le taux de diffusion indépendamment de la direction, et l'anisotropie qui estime la « directionnalité » de la diffusion. Ainsi, une modification de ces indices indique une altération de l'intégrité des fibres de la substance blanche et donc de la communication entre les régions qu'elles relient. Ces mesures permettent d'aboutir à la construction d'une cartographie des différents faisceaux composant la substance blanche (tractographie). La maladie d'Alzheimer entraîne une démyélinisation et une perte axonale. Ces lésions entraînent une diminution de l'anisotropie et une augmentation de la diffusivité dans la substance blanche qui connecte les cortex associatifs (corps calleux, substance blanche des lobes temporaux, frontaux et pariétaux), et au niveau de la substance blanche du système limbique. Ainsi, la majorité des études montrent une atteinte des fibres au niveau du splénium du corps calleux qui connecte les cortex associatifs postérieurs (Rose et coll., 2000 ; Bozzali et coll., 2002 ; Takahashi et coll., 2002 ; Head et coll., 2004 ; Naggara et coll., 2006 ; Zhang et coll., 2007). L'altération de la partie postérieure du cingulum, qui connecte la région hippocampique au cortex cingulaire postérieur, a également été montrée (Takahashi et coll., 2002 ; Yoshiura et coll., 2002), y compris à un stade pré-démentiel (Zhang et coll., 2007). De plus, l'indice d'anisotropie dans cette région serait corrélé au volume de l'hippocampe (Xie et coll., 2005b), ainsi qu'aux performances mnésiques (Fellgiebel et coll., 2005). Les résultats de ces études sont particulièrement intéressants dans la mesure où ils renforcent l'hypothèse d'une dysconnexion entre l'hippocampe et le cortex cingulaire postérieur, avancée pour expliquer l'atteinte fonctionnelle précoce de cette région (voir supra).
). Dans un tissu, la diffusion des molécules d'eau ne se fait pas de façon aléatoire mais elle est contrainte par différents facteurs environnants (les membranes cellulaires, les fibres axonales et diverses macromolécules). Leur diffusion dans la substance blanche, riche en fibres axonales, correspond donc à une diffusion anisotropique qui peut être caractérisée en DTI, grâce à deux indices de cette diffusion anisotropique : la diffusivité qui évalue le taux de diffusion indépendamment de la direction, et l'anisotropie qui estime la « directionnalité » de la diffusion. Ainsi, une modification de ces indices indique une altération de l'intégrité des fibres de la substance blanche et donc de la communication entre les régions qu'elles relient. Ces mesures permettent d'aboutir à la construction d'une cartographie des différents faisceaux composant la substance blanche (tractographie). La maladie d'Alzheimer entraîne une démyélinisation et une perte axonale. Ces lésions entraînent une diminution de l'anisotropie et une augmentation de la diffusivité dans la substance blanche qui connecte les cortex associatifs (corps calleux, substance blanche des lobes temporaux, frontaux et pariétaux), et au niveau de la substance blanche du système limbique. Ainsi, la majorité des études montrent une atteinte des fibres au niveau du splénium du corps calleux qui connecte les cortex associatifs postérieurs (Rose et coll., 2000 ; Bozzali et coll., 2002 ; Takahashi et coll., 2002 ; Head et coll., 2004 ; Naggara et coll., 2006 ; Zhang et coll., 2007). L'altération de la partie postérieure du cingulum, qui connecte la région hippocampique au cortex cingulaire postérieur, a également été montrée (Takahashi et coll., 2002 ; Yoshiura et coll., 2002), y compris à un stade pré-démentiel (Zhang et coll., 2007). De plus, l'indice d'anisotropie dans cette région serait corrélé au volume de l'hippocampe (Xie et coll., 2005b), ainsi qu'aux performances mnésiques (Fellgiebel et coll., 2005). Les résultats de ces études sont particulièrement intéressants dans la mesure où ils renforcent l'hypothèse d'une dysconnexion entre l'hippocampe et le cortex cingulaire postérieur, avancée pour expliquer l'atteinte fonctionnelle précoce de cette région (voir supra).Les résultats au niveau de l'hippocampe sont contradictoires : préservé (Choi et coll., 2005) ou atteint (Fellgiebel et coll., 2004 ; Xie et coll., 2006), mais ils convergent vers une atteinte des faisceaux de la région parahippocampique (Rose et coll., 2006 ; Zhang et coll., 2007), en particulier, de la voie perforante, qui relie le cortex entorhinal à l'hippocampe, cette atteinte étant corrélée avec les scores de mémoire (Kalus et coll., 2006). L'atrophie de la région temporale interne caractéristique de la maladie d'Alzheimer s'accompagne donc d'une vulnérabilité des fibres hippocampiques et parahippocampiques. Les faisceaux hippocampiques sont également atteints chez les patients atteints de MCI (Fellgiebel et coll., 2004 ; Muller et coll., 2005) ainsi que les fibres du cortex parahippocampique (Zhang et coll., 2007), notamment entorhinal (Rose et coll., 2006), la voie perforante en particulier (Kalus et coll., 2006). Là encore, ces atteintes sont corrélées aux faibles performances de rappel mnésique (Muller et coll., 2005 ; Kalus et coll., 2006).
) ou atteint (Fellgiebel et coll., 2004 ; Xie et coll., 2006), mais ils convergent vers une atteinte des faisceaux de la région parahippocampique (Rose et coll., 2006 ; Zhang et coll., 2007), en particulier, de la voie perforante, qui relie le cortex entorhinal à l'hippocampe, cette atteinte étant corrélée avec les scores de mémoire (Kalus et coll., 2006). L'atrophie de la région temporale interne caractéristique de la maladie d'Alzheimer s'accompagne donc d'une vulnérabilité des fibres hippocampiques et parahippocampiques. Les faisceaux hippocampiques sont également atteints chez les patients atteints de MCI (Fellgiebel et coll., 2004 ; Muller et coll., 2005) ainsi que les fibres du cortex parahippocampique (Zhang et coll., 2007), notamment entorhinal (Rose et coll., 2006), la voie perforante en particulier (Kalus et coll., 2006). Là encore, ces atteintes sont corrélées aux faibles performances de rappel mnésique (Muller et coll., 2005 ; Kalus et coll., 2006).Ainsi, la DTI est une technique sensible à des altérations précoces qui ne sont pas mises en évidence en IRM anatomique et qui participeraient aux déficits cognitifs. La DTI pourrait aussi présenter un intérêt diagnostique puisque Kantarci et coll. (2005) ont montré, chez des patients atteints de MCI, que l'indice de diffusivité dans l'hippocampe était un bon indice prédictif de la conversion vers la maladie d'Alzheimer. Enfin, certains résultats obtenus en DTI apportent des arguments qui étayent l'hypothèse d'une dysconnexion entre les régions hippocampique et cingulaire postérieure suggérée par les résultats en imagerie fonctionnelle au repos. La DTI apparaît donc comme une technique complémentaire pour mieux comprendre la physiopathologie de la maladie d'Alzheimer.
) ont montré, chez des patients atteints de MCI, que l'indice de diffusivité dans l'hippocampe était un bon indice prédictif de la conversion vers la maladie d'Alzheimer. Enfin, certains résultats obtenus en DTI apportent des arguments qui étayent l'hypothèse d'une dysconnexion entre les régions hippocampique et cingulaire postérieure suggérée par les résultats en imagerie fonctionnelle au repos. La DTI apparaît donc comme une technique complémentaire pour mieux comprendre la physiopathologie de la maladie d'Alzheimer.Imagerie moléculaire
De nouveaux marqueurs TEP permettant le marquage in vivo des atteintes neuropathologiques (DNF et/ou plaques séniles) ont été récemment développés. Plusieurs études ont ainsi été réalisées chez l'homme (Shoghi-Jadid et coll., 2002 ; Klunk et coll., 2004 ; Small et coll., 2006). Klunk et collaborateurs (2004) ont réalisé une étude en TEP avec un nouveau traceur, le 11C-PIB, qui marque de façon relativement sélective les dépôts β-amyloïdes. Leurs résultats et ceux d'autres études convergent vers une localisation préférentielle de ce traceur dans les régions corticales, surtout frontales, temporo-pariétales et cingulaires postérieures, en contraste avec une faible localisation dans la région hippocampique (Kemppainen et coll., 2006 ; Edison et coll., 2007). Selon l'étude récente de Ziolko et coll. (2006), les mesures de rétention du PIB seraient plus efficaces que celles de la consommation de glucose pour différencier un groupe de patients atteints de maladie d'Alzheimer et un groupe de sujets sains. Dans la première étude longitudinale réalisée à l'aide de cette technique, Engler et coll. (2006) ont montré qu'alors que la consommation cérébrale de glucose a diminué d'environ 20 % entre deux examens TEP-FDG réalisés à deux ans d'intervalle, le taux de rétention du PIB n'a pas augmenté, suggérant l'existence d'une phase de plateau assez tôt dans l'évolution de la maladie. Cette étude montre également des corrélations significatives avec des performances mnésiques, dans le sens attendu, positives avec la consommation de glucose et négatives avec le taux de rétention du PIB (voir aussi Edison et coll., 2007). Selon Klunk et collaborateurs (2006), ces résultats pourraient cependant être influencés par la progression du taux d'atrophie, qui accentue la diminution de la consommation de glucose alors qu'elle aurait plutôt tendance à diminuer le taux de rétention du PIB. De plus, il existe une importante variabilité inter-individuelle, qui ne permet pas de généraliser ces résultats. En effet, la rétention du PIB a augmenté chez plus de la moitié des patients, mais ceci est masqué par les résultats moyens du groupe. Deux publications portant sur un petit groupe de patients atteints de MCI (dont plusieurs sont les mêmes) montrent que certains patients se comportent comme des sujets sains et d'autres comme des patients atteints de maladie Alzheimer (Lopresti et coll., 2005 ; Price et coll., 2005). Mintun et coll. (2006) ont de plus mis en évidence des anomalies de la rétention du PIB chez quelques sujets sains, ce qui, selon eux, pourrait augurer une évolution ultérieure vers la maladie d'Alzheimer, qui reste toutefois à confirmer. Ainsi, ce marqueur présente un intérêt majeur dans l'étude de la maladie d'Alzheimer, y compris à un stade très précoce, même s'il est limité par son absence de sensibilité pour l'atteinte de la région hippocampique, épargnée par les dépôts amyloïdes au début de la maladie (De Leon et Klunk, 2006, pour revue).
; Klunk et coll., 2004 ; Small et coll., 2006). Klunk et collaborateurs (2004) ont réalisé une étude en TEP avec un nouveau traceur, le 11C-PIB, qui marque de façon relativement sélective les dépôts β-amyloïdes. Leurs résultats et ceux d'autres études convergent vers une localisation préférentielle de ce traceur dans les régions corticales, surtout frontales, temporo-pariétales et cingulaires postérieures, en contraste avec une faible localisation dans la région hippocampique (Kemppainen et coll., 2006 ; Edison et coll., 2007). Selon l'étude récente de Ziolko et coll. (2006), les mesures de rétention du PIB seraient plus efficaces que celles de la consommation de glucose pour différencier un groupe de patients atteints de maladie d'Alzheimer et un groupe de sujets sains. Dans la première étude longitudinale réalisée à l'aide de cette technique, Engler et coll. (2006) ont montré qu'alors que la consommation cérébrale de glucose a diminué d'environ 20 % entre deux examens TEP-FDG réalisés à deux ans d'intervalle, le taux de rétention du PIB n'a pas augmenté, suggérant l'existence d'une phase de plateau assez tôt dans l'évolution de la maladie. Cette étude montre également des corrélations significatives avec des performances mnésiques, dans le sens attendu, positives avec la consommation de glucose et négatives avec le taux de rétention du PIB (voir aussi Edison et coll., 2007). Selon Klunk et collaborateurs (2006), ces résultats pourraient cependant être influencés par la progression du taux d'atrophie, qui accentue la diminution de la consommation de glucose alors qu'elle aurait plutôt tendance à diminuer le taux de rétention du PIB. De plus, il existe une importante variabilité inter-individuelle, qui ne permet pas de généraliser ces résultats. En effet, la rétention du PIB a augmenté chez plus de la moitié des patients, mais ceci est masqué par les résultats moyens du groupe. Deux publications portant sur un petit groupe de patients atteints de MCI (dont plusieurs sont les mêmes) montrent que certains patients se comportent comme des sujets sains et d'autres comme des patients atteints de maladie Alzheimer (Lopresti et coll., 2005 ; Price et coll., 2005). Mintun et coll. (2006) ont de plus mis en évidence des anomalies de la rétention du PIB chez quelques sujets sains, ce qui, selon eux, pourrait augurer une évolution ultérieure vers la maladie d'Alzheimer, qui reste toutefois à confirmer. Ainsi, ce marqueur présente un intérêt majeur dans l'étude de la maladie d'Alzheimer, y compris à un stade très précoce, même s'il est limité par son absence de sensibilité pour l'atteinte de la région hippocampique, épargnée par les dépôts amyloïdes au début de la maladie (De Leon et Klunk, 2006, pour revue).Shoghi-Jadid et coll. (2002) ont utilisé un autre traceur, le 18F-FDDNP, qui marque à la fois les DNF et les dépôts β-amyloïdes. Les auteurs montrent une accumulation importante de ce marqueur dans la région hippocampique, une bonne superposition des zones de fixation du [18F]FDDNP et des zones d'hypométabolisme mises en évidence par le 18FDG ainsi qu'une corrélation négative entre l'accumulation de ce marqueur et le degré global d'atteinte cognitive, ainsi que des troubles de la mémoire épisodique. La localisation préférentielle de ce marqueur ainsi que son lien avec les troubles cognitifs suggèrent qu'il reflète au moins la pathologie Tau (Lockhart, 2006). Une étude récente publiée par la même équipe (Small et coll., 2006) montre que ce marqueur est plus puissant que le FDG pour différentier des groupes de patients atteints de MCI ou de maladie d'Alzheimer et des sujets témoins. Ces résultats prometteurs laissent entrevoir l'intérêt de ces marqueurs non seulement pour la compréhension de la physiopathologie de la maladie d'Alzheimer, mais aussi dans le domaine de la recherche thérapeutique.
) ont utilisé un autre traceur, le 18F-FDDNP, qui marque à la fois les DNF et les dépôts β-amyloïdes. Les auteurs montrent une accumulation importante de ce marqueur dans la région hippocampique, une bonne superposition des zones de fixation du [18F]FDDNP et des zones d'hypométabolisme mises en évidence par le 18FDG ainsi qu'une corrélation négative entre l'accumulation de ce marqueur et le degré global d'atteinte cognitive, ainsi que des troubles de la mémoire épisodique. La localisation préférentielle de ce marqueur ainsi que son lien avec les troubles cognitifs suggèrent qu'il reflète au moins la pathologie Tau (Lockhart, 2006). Une étude récente publiée par la même équipe (Small et coll., 2006) montre que ce marqueur est plus puissant que le FDG pour différentier des groupes de patients atteints de MCI ou de maladie d'Alzheimer et des sujets témoins. Ces résultats prometteurs laissent entrevoir l'intérêt de ces marqueurs non seulement pour la compréhension de la physiopathologie de la maladie d'Alzheimer, mais aussi dans le domaine de la recherche thérapeutique.En conclusion,
les différentes méthodes d'imagerie cérébrale ont conduit à une meilleure compréhension de la physiopathologie de la maladie d'Alzheimer et des liens entre anomalies cérébrales et expression clinique. L'imagerie cérébrale fonctionnelle est la plus puissante en terme de diagnostic précoce et de prédiction du déclin cognitif. Même si les résultats obtenus à partir de groupes de patients ne sont pas encore applicables au niveau individuel, ils ont mis l'accent sur des anomalies qui peuvent faire craindre un pronostic péjoratif. L'imagerie apparaît supérieure aux autres méthodes (Chételat et coll., 2005b ; Zamrini et coll., 2004), mais la combinaison des approches d'imagerie et de neuropsychologie est recommandée car elle s'avère plus efficace que le recours à une seule technique (Kitayama et coll., 2001 ; Huang et coll., 2003 ; Cabranes et coll., 2004 ; Chong et Sahadevan, 2005, pour revue). Il serait cependant inutile de multiplier les mesures. Les indices les plus efficaces sont l'atrophie de la région hippocampique (s'étendant à une autre région), l'hypométabolisme du cortex cingulaire postérieur et temporo-pariétal associés à des troubles « authentiques » de la mémoire épisodique.
; Zamrini et coll., 2004), mais la combinaison des approches d'imagerie et de neuropsychologie est recommandée car elle s'avère plus efficace que le recours à une seule technique (Kitayama et coll., 2001 ; Huang et coll., 2003 ; Cabranes et coll., 2004 ; Chong et Sahadevan, 2005, pour revue). Il serait cependant inutile de multiplier les mesures. Les indices les plus efficaces sont l'atrophie de la région hippocampique (s'étendant à une autre région), l'hypométabolisme du cortex cingulaire postérieur et temporo-pariétal associés à des troubles « authentiques » de la mémoire épisodique.De nouvelles méthodes d'imagerie se développent actuellement pour mettre en évidence les troubles de la connectivité entre les régions et les anomalies de la substance blanche, et pour étudier in vivo la distribution régionale des plaques séniles et des dégénérescences neurofibrillaires, mais leur application clinique n'est pas immédiate. Des études, réalisées en électrophysiologie, ont montré des anomalies du tracé EEG (Koenig et coll., 2005 ; Babiloni et coll., 2006), ou des potentiels évoqués (Olichney et coll., 2002 ; Phillips et coll., 2004 ; Missonier et coll., 2005 ; Olichney et coll., 2006). D'autres études ont utilisé la spectroscopie pour détecter des perturbations du fonctionnement neuronal (Norfray et Provenzale, 2004, pour revue ; Modrego et coll., 2005), y compris dans le MCI (Ackl et coll., 2005). Les résultats sont cependant difficiles à appliquer au niveau individuel, car non spécifiques.
; Babiloni et coll., 2006), ou des potentiels évoqués (Olichney et coll., 2002 ; Phillips et coll., 2004 ; Missonier et coll., 2005 ; Olichney et coll., 2006). D'autres études ont utilisé la spectroscopie pour détecter des perturbations du fonctionnement neuronal (Norfray et Provenzale, 2004, pour revue ; Modrego et coll., 2005), y compris dans le MCI (Ackl et coll., 2005). Les résultats sont cependant difficiles à appliquer au niveau individuel, car non spécifiques.De nombreux articles concluent sur les perspectives d'application au diagnostic précoce, quelle que soit la méthode d'imagerie employée, notamment l'IRMf, et même l'imagerie moléculaire. Quelques études suggèrent même la possibilité de détecter des anomalies cérébrales chez des sujets sains susceptibles de répondre ensuite aux critères de MCI (De Leon et Klunk, 2006, pour revue). Cependant, compte tenu des contraintes expérimentales imposées par de tels protocoles (élaboration du paradigme, groupe témoins, coûts...), il est difficile d'imaginer leur faisabilité en routine. À l'heure actuelle, c'est l'imagerie morphologique en IRM qui reste la plus facile à mettre en œ uvre. Sa contribution devrait encore s'intensifier avec l'automatisation de techniques de traitement d'image permettant de visualiser l'atrophie cérébrale dans des régions jouant un rôle crucial dans la survenue des troubles cognitifs. Parmi les autres méthodes, la TEP pourrait jouer un rôle important sur le plan clinique dans les années à venir, en particulier en cas de diagnostic difficile.
, pour revue). Cependant, compte tenu des contraintes expérimentales imposées par de tels protocoles (élaboration du paradigme, groupe témoins, coûts...), il est difficile d'imaginer leur faisabilité en routine. À l'heure actuelle, c'est l'imagerie morphologique en IRM qui reste la plus facile à mettre en œ uvre. Sa contribution devrait encore s'intensifier avec l'automatisation de techniques de traitement d'image permettant de visualiser l'atrophie cérébrale dans des régions jouant un rôle crucial dans la survenue des troubles cognitifs. Parmi les autres méthodes, la TEP pourrait jouer un rôle important sur le plan clinique dans les années à venir, en particulier en cas de diagnostic difficile.Bibliographie
[1] ackl n, ising m, schreiber ya, atiya m, sonntag a, auer dp. Hippocampal metabolic abnormalities in mild cognitive impairment and Alzheimer’s disease.
Neurosci Lett. 2005;
384:23- 28
[2] alexander ge, chen k, pietrini p, rapoport si, reiman em. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies.
Am J Psychiatry. 2002;
159:738- 745
[3] anchisi d, borroni b, franceschi m, kerrouche n, kalbe e. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease.
Arch Neurol. 2005;
62:1728- 1733
[4] apostolova lg, dutton ra, dinov id, hayashi km, toga aw. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps.
Arch Neurol. 2006;
63:693- 699
[5] arnaiz e, jelic v, almkvist o, wahlund lo, winblad b, valind s. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment.
Neuroreport. 2001;
12:851- 855
[6] babiloni c, binetti g, cassetta e, dal forno g, del percio c, ferreri f. Sources of cortical rhythms change as a function of cognitive impairment in pathological aging: a multicenter study.
Clin Neurophysiol. 2006;
117:252- 268
[7] backman l, andersson jl, nyberg l, winblad b, nordberg a, almkvist o. Brain regions associated with episodic retrieval in normal aging and Alzheimer’s disease.
Neurology. 1999;
52:1861- 1870
[8] baron jc, chetelat g, desgranges b, perchey g, landeau b. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer’s disease.
Neuroimage. 2001;
14:298- 309
[9] bassett ss, yousem dm, cristinzio c, kusevic i, yassa ma. Familial risk for Alzheimer’s disease alters fMRI activation patterns.
Brain. 2006;
129:1229- 1239
[10] bastos leite aj, scheltens p, barkhof f. Pathological aging of the brain: an overview.
Top Magn Reson Imaging. 2004;
15:369- 389
[11] becker jt, mintun ma, aleva k, wiseman mb, nichols t, dekosky st. Compensatory reallocation of brain resources supporting verbal episodic memory in Alzheimer’s disease.
Neurology. 1996;
46:692- 700
[12] bokde al, pietrini p, ibanez v, furey ml, alexander ge, graff-radford nr. The effect of brain atrophy on cerebral hypometabolism in the visual variant of Alzheimer disease.
Arch Neurol. 2001;
58:480- 486
[13] bondi mw, houston ws, eyler lt, brown gg. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease.
Neurology. 2005;
64:501- 508
[14] bookheimer sy, strojwas mh, cohen ms, saunders am, pericak-vance ma. Patterns of brain activation in people at risk for Alzheimer’s disease.
N Engl J Med. 2000;
343:450- 456
[15] boxer al, rankin kp, miller bl, schuff n, weiner m. Cinguloparietal atrophy distinguishes Alzheimer disease from semantic dementia.
Arch Neurol. 2003;
60:949- 956
[16] bozzali m, falini a, franceschi m, cercignani m, zuffi m. White matter damage in Alzheimer’s disease assessed in vivo using diffusion tensor magnetic resonance imaging.
J Neurol Neurosurg Psychiatry. 2002;
72:742- 746
[17] bozzali m, filippi m, magnani g, cercignani m, franceschi m. The contribution of voxel-based morphometry in staging patients with mild cognitive impairment.
Neurology. 2006;
67:453- 460
[18] buckner rl, snyder az, shannon bj, larossa g, sachs r. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory.
J Neurosci. 2005;
25:7709- 7717
[19] burton ej, karas g, paling sm, barber r, williams ed. Patterns of cerebral atrophy in dementia with Lewy bodies using voxel-based morphometry.
Neuroimage. 2002;
17:618- 630
[20] busatto gf, garrido ge, almeida op, castro cc, camargo ch. A voxel-based morphometry study of temporal lobe gray matter reductions in Alzheimer’s disease.
Neurobiol Aging. 2003;
24:221- 231
[21] cabeza r, nyberg l. Imaging cognition II: An empirical review of 275 PET and fMRI studies.
J Cogn Neurosci. 2000;
12:1- 47
[22] cabranes ja, de juan r, encinas m, marcos a, gil p. Relevance of functional neuroimaging in the progression of mild cognitive impairment.
Neurol Res. 2004;
26:496- 501
[23] celone ka, calhoun vd, dickerson bc, atri a, chua ef. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: an independent component analysis.
J Neurosci. 2006;
26:10222- 10231
[24] chetelat g, baron jc. Early diagnosis of Alzheimer’s disease: contribution of structural neuroimaging.
Neuroimage. 2003;
18:525- 541
[25] chetelat g, desgranges b, de la sayette v, viader f, eustache f, baron jc. Mapping gray matter loss with voxel-based morphometry in mild cognitive impairment.
Neuroreport. 2002;
13:1939- 1943
[26] chetelat g, desgranges b, de la sayette v, viader f, eustache f, baron jc. Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease?.
Neurology. 2003a;
60:1374- 1377
[27] chetelat g, desgranges b, de la sayette v, viader f, berkouk k. Dissociating atrophy and hypometabolism impact on episodic memory in mild cognitive impairment.
Brain. 2003b;
126:1955- 1967
[28] chetelat g, landeau b, eustache f, mezenge f, viader f. Using voxel-based morphometry to map the structural changes associated with rapid conversion in MCI: a longitudinal MRI study.
Neuroimage. 2005a;
27:934- 946
[29] chetelat g, eustache f, viader f, de la sayette v, pelerin a. FDG-PET measurement is more accurate than neuropsychological assessments to predict global cognitive deterioration in patients with mild cognitive impairment.
Neurocase. 2005b;
11:14- 25
[30] chetelat g, desgranges b, eustache f. Brain profile of hypometabolism in early Alzheimer’s disease: relationships with cognitive deficits and atrophy.
Rev Neurol (Paris). 2006;
162:945- 951
[31] choi sj, lim ko, monteiro i, reisberg b. Diffusion tensor imaging of frontal white matter microstructure in early Alzheimer’s disease: a preliminary study.
J Geriatr Psychiatry Neurol. 2005;
18:12- 19
[32] chong ms, sahadevan s. Preclinical Alzheimer’s disease: diagnosis and prediction of progression.
Lancet Neurol. 2005;
4:576- 579
[33] csernansky jg, wang l, swank j, miller jp, gado m. Preclinical detection of Alzheimer’s disease: hippocampal shape and volume predict dementia onset in the elderly.
Neuroimage. 2005;
25:783- 792
[34] d’esposito m, deouell ly, gazzaley a. Alterations in the BOLD fMRI signal with ageing and disease: a challenge for neuroimaging.
Nat Rev Neurosci. 2003;
4:863- 872
[35] de leon mj, klunk w. Biomarkers for the early diagnosis of Alzheimer’s disease.
Lancet Neurol. 2006;
5:198- 199
[36] dekosky st, ikonomovic md, styren sd, beckett l, wisniewski s. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment.
Ann Neurol. 2002;
51:145- 155
[37] delacourte a, david jp, sergeant n, buee l, wattez a. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease.
Neurology. 1999;
52:1158- 1165
[38] demetriades ak. Functional neuroimaging in Alzheimer’s type dementia.
J Neurol Sci. 2002;
203-204:247- 251
[39] desgranges b, baron jc, eustache f. The functional neuroanatomy of episodic memory: the role of the frontal lobes, the hippocampal formation, and other areas.
Neuroimage. 1998a;
8:198- 213
[40] desgranges b, baron jc, de la sayette v, petit-taboue mc, benali k. The neural substrates of memory systems impairment in Alzheimer’s disease. A PET study of resting brain glucose utilization.
Brain. 1998b;
121:611- 631
[41] desgranges b, baron jc, lalevee c, giffard b, viader f. The neural substrates of episodic memory impairment in Alzheimer’s disease as revealed by FDG-PET: relationship to degree of deterioration.
Brain. 2002;
125:1116- 1124
[42] desgranges b, chetelat g, eustache f. Brain substrates of episodic memory disorders in Alzheimer’s disease.
Rev Neurol (Paris). 2004;
160:S44- S54
[43] desgranges b, eustache f. Neuroanatomie fonctionnelle des troubles de la mémoire dans la maldie d’Alzheimer.
In: ergis a, gely-nargeot m, van der linden m, editors.
Les troubles de la mémoire dans la maladie d’Alzheimer..
Solal:Marseille;
2005.
p. 297- 322
[44] devous md sr. Functional brain imaging in the dementias: role in early detection, differential diagnosis, and longitudinal studies.
Eur J Nucl Med Mol Imaging. 2002;
29:1685- 1696
[45] deweer b, lehericy s, pillon b, baulac m, chiras j. Memory disorders in probable Alzheimer’s disease: the role of hippocampal atrophy as shown with MRI.
J Neurol Neurosurg Psychiatry. 1995;
58:590- 597
[46] dickerson bc, salat dh, bates jf, atiya m, killiany rj. Medial temporal lobe function and structure in mild cognitive impairment.
Ann Neurol. 2004;
56:27- 35
[47] dickerson bc, salat dh, greve dn, chua ef, rand-giovannetti e. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD.
Neurology. 2005;
65:404- 411
[48] drzezga a, lautenschlager n, siebner h, riemenschneider m, willoch f. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study.
Eur J Nucl Med Mol Imaging. 2003;
30:1104- 1113
[49] du at, schuff n, amend d, laakso mp, hsu yy. Magnetic resonance imaging of the entorhinal cortex and hippocampus in mild cognitive impairment and Alzheimer’s disease.
J Neurol Neurosurg Psychiatry. 2001;
71:441- 447
[50] duyckaerts c, colle ma, dessi f, grignon y, piette f, hauw jj. The progression of the lesions in Alzheimer disease: insights from a prospective clinicopathological study.
J Neural Transm Suppl. 1998;
53:119- 126
[51] edison p, archer ha, hinz r, hammers a, pavese n. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study.
Neurology. 2007;
68:501- 508
[52] elgh e, larsson a, eriksson s, nyberg l. Altered prefrontal brain activity in persons at risk for Alzheimer’s disease: an fMRI study.
Int Psychogeriatr. 2003;
15:121- 133
[53] engler h, forsberg a, almkvist o, blomquist g, larsson e. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease.
Brain. 2006;
129:2856- 2866
[54] eustache f, desgranges b, giffard b, de la sayette v, baron jc. Entorhinal cortex disruption causes memory deficit in early Alzheimer’s disease as shown by PET.
Neuroreport. 2001;
12:683- 685
[55] eustache f, piolino p, giffard b, viader f, de la sayette v. ’In the course of time’: a PET study of the cerebral substrates of autobiographical amnesia in Alzheimer’s disease.
Brain. 2004;
127:1549- 1560
[56] fellgiebel a, wille p, muller mj, winterer g, scheurich a. Ultrastructural hippocampal and white matter alterations in mild cognitive impairment: a diffusion tensor imaging study.
Dement Geriatr Cogn Disord. 2004;
18:101- 108
[57] fellgiebel a, muller mj, wille p, dellani pr, scheurich a. Color-coded diffusion-tensor-imaging of posterior cingulate fiber tracts in mild cognitive impairment.
Neurobiol Aging. 2005;
26:1193- 1198
[58] fleisher as, houston ws, eyler lt, frye s, jenkins c. Identification of Alzheimer disease risk by functional magnetic resonance imaging.
Arch Neurol. 2005;
62:1881- 1888
[59] fouquet m, villain n, chetelat g, eustache f, desgranges b. Imagerie cérébrale et physiopathologie de la maladie d’Alzheimer.
Psychol NeuroPsychiatr Vieil. 2007, sous presse;
:-
[60] frisoni gb, testa c, zorzan a, sabattoli f, beltramello a. Detection of grey matter loss in mild Alzheimer’s disease with voxel based morphometry.
J Neurol Neurosurg Psychiatry. 2002;
73:657- 664
[61] garrido ge, furuie ss, buchpiguel ca, bottino cm, almeida op. Relation between medial temporal atrophy and functional brain activity during memory processing in Alzheimer’s disease: a combined MRI and SPECT study.
J Neurol Neurosurg Psychiatry. 2002;
73:508- 516
[62] gee j, ding l, xie z, lin m, devita c, grossman m. Alzheimer’s disease and frontotemporal dementia exhibit distinct atrophy-behavior correlates: a computer-assisted imaging study.
Acad Radiol. 2003;
10:1392- 1401
[63] gilboa a, ramirez j, kohler s, westmacott r, black se, moscovitch m. Retrieval of autobiographical memory in Alzheimer’s disease: relation to volumes of medial temporal lobe and other structures.
Hippocampus. 2005;
15:535- 550
[64] golby a, silverberg g, race e, gabrieli s, o’shea j. Memory encoding in Alzheimer’s disease: an fMRI study of explicit and implicit memory.
Brain. 2005;
128:773- 787
[65] good cd, scahill ri, fox nc, ashburner j, friston kj. Automatic differentiation of anatomical patterns in the human brain: validation with studies of degenerative dementias.
Neuroimage. 2002;
17:29- 46
[66] gould rl, brown rg, owen am, bullmore et, williams sc, howard rj. Functional neuroanatomy of successful paired associate learning in Alzheimer’s disease.
Am J Psychiatry. 2005;
162:2049- 2060
[67] grady cl, haxby jv, horwitz b, gillette j, salerno ja. Activation of cerebral blood flow during a visuoperceptual task in patients with Alzheimer-type dementia.
Neurobiol Aging. 1993;
14:35- 44
[68] grady cl, mcintosh ar, beig s, keightley ml, burian h, black se. Evidence from functional neuroimaging of a compensatory prefrontal network in Alzheimer’s disease.
J Neurosci. 2003;
23:986- 993
[69] greicius md, srivastava g, reiss al, menon v. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI.
Proc Natl Acad Sci USA. 2004;
101:4637- 4642
[70] gron g, bittner d, schmitz b, wunderlich ap, riepe mw. Subjective memory complaints: objective neural markers in patients with Alzheimer’s disease and major depressive disorder.
Ann Neurol. 2002;
51:491- 498
[71] gron g, riepe mw. Neural basis for the cognitive continuum in episodic memory from health to Alzheimer disease.
Am J Geriatr Psychiatry. 2004;
12:648- 652
[72] grossman m, koenig p, glosser g, devita c, moore p. Neural basis for semantic memory difficulty in Alzheimer’s disease: an fMRI study.
Brain. 2003;
126:292- 311
[73] grossman m, mcmillan c, moore p, ding l, glosser g. What’s in a name: voxel-based morphometric analyses of MRI and naming difficulty in Alzheimer’s disease, frontotemporal dementia and corticobasal degeneration.
Brain. 2004;
127:628- 649
[74] gusnard da, raichle me, raichle me. Searching for a baseline: functional imaging and the resting human brain.
Nat Rev Neurosci. 2001;
2:685- 694
[75] hamalainen a, pihlajamaki m, tanila h, hanninen t, niskanen e. Increased fMRI responses during encoding in mild cognitive impairment.
Neurobiol Aging. sous presse;
:-
[76] hampel h, teipel sj, bayer w, alexander ge, schwarz r. Age transformation of combined hippocampus and amygdala volume improves diagnostic accuracy in Alzheimer’s disease.
J Neurol Sci. 2002;
194:15- 19
[77] han sd, houston ws, jak aj, eyler lt, nagel bj. Verbal paired-associate learning by APOE genotype in non-demented older adults: fMRI evidence of a right hemispheric compensatory response.
Neurobiol Aging. 2007;
28:238- 247
[78] hao j, li k, li k, zhang d, wang w. Visual attention deficits in Alzheimer’s disease: an fMRI study.
Neurosci Lett. 2005;
385:18- 23
[79] head d, buckner rl, shimony js, williams le, akbudak e. Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the Alzheimer type: evidence from diffusion tensor imaging.
Cereb Cortex. 2004;
14:410- 423
[80] herholz k, salmon e, perani d, baron jc, holthoff v. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET.
Neuroimage. 2002;
17:302- 316
[81] herholz k. PET studies in dementia.
Ann Nucl Med. 2003;
17:79- 89
[82] heun r, freymann k, erb m, leube dt, jessen f. Mild cognitive impairment (MCI) and actual retrieval performance affect cerebral activation in the elderly.
Neurobiol Aging. 2007;
28:404- 413
[83] hirao k, ohnishi t, hirata y, yamashita f, mori t. The prediction of rapid conversion to Alzheimer’s disease in mild cognitive impairment using regional cerebral blood flow SPECT.
Neuroimage. 2005;
28:1014- 1021
[84] hirata y, matsuda h, nemoto k, ohnishi t, hirao k. Voxel-based morphometry to discriminate early Alzheimer’s disease from controls.
Neurosci Lett. 2005;
382:269- 274
[85] hirono n, mori e, ishii k, imamura t, shimomura t. Regional metabolism: associations with dyscalculia in Alzheimer’s disease.
J Neurol Neurosurg Psychiatry. 1998;
65:913- 916
[86] hirono n, mori e, ishii k, imamura t, tanimukai s. Neuronal substrates for semantic memory: a positron emission tomography study in Alzheimer’s disease.
Dement Geriatr Cogn Disord. 2001;
12:15- 21
[87] hirono n, hashimoto m, yasuda m, ishii k, sakamoto s. The effect of APOE epsilon4 allele on cerebral glucose metabolism in AD is a function of age at onset.
Neurology. 2002;
58:743- 750
[88] huang c, wahlund lo, svensson l, winblad b, julin p. Cingulate cortex hypoperfusion predicts Alzheimer’s disease in mild cognitive impairment.
BMC Neurol. 2002;
2:9
[89] huang c, wahlund lo, almkvist o, elehu d, svensson l. Voxel- and VOI-based analysis of SPECT CBF in relation to clinical and psychological heterogeneity of mild cognitive impairment.
Neuroimage. 2003;
19:1137- 1144
[90] iadecola c. Neurovascular regulation in the normal brain and in Alzheimer’s disease.
Nat Rev Neurosci. 2004;
5:347- 360
[91] ibanez v, pietrini p, alexander ge, furey ml, teichberg d. Regional glucose metabolic abnormalities are not the result of atrophy in Alzheimer’s disease.
Neurology. 1998;
50:1585- 1593
[92] ishii k, sasaki m, yamaji s, sakamoto s, kitagaki h, mori e. Paradoxical hippocampus perfusion in mild-to-moderate Alzheimer’s disease.
J Nucl Med. 1998;
39:293- 298
[93] jin k, peel al, mao xo, xie l, cottrell ba. Increased hippocampal neurogenesis in Alzheimer’s disease.
Proc Natl Acad Sci USA. 2004;
101:343- 347
[94] johnson sc, saykin aj, baxter lc, flashman la, santulli rb. The relationship between fMRI activation and cerebral atrophy: comparison of normal aging and alzheimer disease.
Neuroimage. 2000;
11:179- 187
[95] johnson sc, schmitz tw, moritz ch, meyerand me, rowley ha. Activation of brain regions vulnerable to Alzheimer’s disease: the effect of mild cognitive impairment.
Neurobiol Aging. 2006a;
27:1604- 1612
[96] johnson sc, schmitz tw, trivedi ma, ries ml, torgerson bm. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation.
J Neurosci. 2006b;
26:6069- 6076
[97] kalpouzos g, eustache f, de la sayette v, viader f, chetelat g, desgranges b. Working memory and FDG-PET dissociate early and late onset Alzheimer disease patients.
J Neurol. 2005;
252:548- 558
[98] kalus p, slotboom j, gallinat j, mahlberg r, cattapan-ludewig k, wiest r. Examining the gateway to the limbic system with diffusion tensor imaging: the perforant pathway in dementia.
Neuroimage. 2006;
30:713- 720
[99] kantarci k, petersen rc, boeve bf, knopman ds, weigand sd. DWI predicts future progression to Alzheimer disease in amnestic mild cognitive impairment.
Neurology. 2005;
64:902- 904
[100] karas gb, burton ej, rombouts sa, van schijndel ra, o’brien jt. A comprehensive study of gray matter loss in patients with Alzheimer’s disease using optimized voxel-based morphometry.
Neuroimage. 2003;
18:895- 907
[101] karas gb, scheltens p, rombouts sa, visser pj, van schijndel ra. Global and local gray matter loss in mild cognitive impairment and Alzheimer’s disease.
Neuroimage. 2004;
23:708- 716
[102] kato t, knopman d, liu h. Dissociation of regional activation in mild AD during visual encoding: a functional MRI study.
Neurology. 2001;
57:812- 816
[103] kawachi t, ishii k, sakamoto s, sasaki m, mori t. Comparison of the diagnostic performance of FDG-PET and VBM-MRI in very mild Alzheimer’s disease.
Eur J Nucl Med Mol Imaging. 2006;
33:801- 809
[104] kemppainen nm, aalto s, wilson ia, nagren k, helin s. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease.
Neurology. 2006;
67:1575- 1580
[105] killiany rj, gomez-isla t, moss m, kikinis r, sandor t. Use of structural magnetic resonance imaging to predict who will get Alzheimer’s disease.
Ann Neurol. 2000;
47:430- 439
[106] kim ej, cho ss, jeong y, park kc, kang sj. Glucose metabolism in early onset versus late onset Alzheimer’s disease: an SPM analysis of 120 patients.
Brain. 2005;
128:1790- 1801
[107] kircher t, weis s, freymann k, erb m, jessen f. Hippocampal activation in MCI patients is necessary for successful memory encoding.
J Neurol Neurosurg Psychiatry. 2007, sous presse;
:-
[108] kitayama n, matsuda h, ohnishi t, kogure d, asada t. Measurements of both hippocampal blood flow and hippocampal gray matter volume in the same individuals with Alzheimer’s disease.
Nucl Med Commun. 2001;
22:473- 477
[109] klunk we, engler h, nordberg a, wang y, blomqvist g. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B.
Ann Neurol. 2004;
55:306- 319
[110] klunk we, mathis ca, price jc, lopresti bj, dekosky st. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease.
Brain. 2006;
129:2805- 2807
[111] koenig t, prichep l, dierks t, hubl d, wahlund lo. Decreased EEG synchronization in Alzheimer’s disease and mild cognitive impairment.
Neurobiol Aging. 2005;
26:165- 171
[112] kogure d, matsuda h, ohnishi t, asada t, uno m. Longitudinal evaluation of early Alzheimer’s disease using brain perfusion SPECT.
J Nucl Med. 2000;
41:1155- 1162
[113] korf es, wahlund lo, visser pj, scheltens p. Medial temporal lobe atrophy on MRI predicts dementia in patients with mild cognitive impairment.
Neurology. 2004;
63:94- 100
[114] le bihan d, mangin jf, poupon c, clark ca, pappata s. Diffusion tensor imaging: concepts and applications.
J Magn Reson Imaging. 2001;
13:534- 546
[115] lehericy s, baulac m, chiras j, pierot l, martin n. Amygdalohippocampal MR volume measurements in the early stages of Alzheimer disease.
AJNR Am J Neuroradiol. 1994;
15:929- 937
[116] lekeu f, van der linden m, degueldre c, lemaire c, luxen a. Effects of Alzheimer’s disease on the recognition of novel versus familiar words: neuropsychological and clinico-metabolic data.
Neuropsychology. 2003;
17:143- 154
[117] lind j, persson j, ingvar m, larsson a, cruts m. Reduced functional brain activity response in cognitively intact apolipoprotein E epsilon4 carriers.
Brain. 2006;
129:1240- 1248
[118] lockhart a. Imaging Alzheimer’s disease pathology: one target, many ligands.
Drug Discov Today. 2006;
11:1093- 1099
[119] lopresti bj, klunk we, mathis ca, hoge ja, ziolko sk. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis.
J Nucl Med. 2005;
46:1959- 1972
[120] machulda mm, ward ha, borowski b, gunter jl, cha rh. Comparison of memory fMRI response among normal, MCI, and Alzheimer’s patients.
Neurology. 2003;
61:500- 506
[121] matsuda h. Cerebral blood flow and metabolic abnormalities in Alzheimer’s disease.
Ann Nucl Med. 2001;
15:85- 92
[122] matsuda h, kanetaka h, ohnishi t, asada t, imabayashi e. Brain SPET abnormalities in Alzheimer’s disease before and after atrophy correction.
Eur J Nucl Med Mol Imaging. 2002;
9:1502- 1505
[123] meguro k, blaizot x, kondoh y, le mestric c, baron jc, chavoix c. Neocortical and hippocampal glucose hypometabolism following neurotoxic lesions of the entorhinal and perirhinal cortices in the non-human primate as shown by PET. Implications for Alzheimer’s disease.
Brain. 1999;
122:1519- 1531
[124] meguro k, lemestric c, landeau b, desgranges b, eustache f, baron jc. Relations between hypometabolism in the posterior association neocortex and hippocampal atrophy in Alzheimer’s disease: a PET/MRI correlative study.
J Neurol Neurosurg Psychiatry. 2001;
71:315- 321
[125] mevel k, desgranges b, baron jc, landeau b, de la sayette v. Detecting hippocampal hypometabolism in Mild Cognitive Impairment using automatic voxel-based approaches.
NeuroImage. sous presse;
:-
[126] minoshima s, giordani b, berent s, frey ka, foster nl, kuhl de. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease.
Ann Neurol. 1997;
42:85- 94
[127] mintun ma, larossa gn, sheline yi, dence cs, lee sy, mach rh. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease.
Neurology. 2006;
67:446- 452
[128] missonnier p, gold g, fazio-costa l, michel j, mulligan r. Early event-related potential changes during working memory activation predict rapid decline in mild cognitive impairment.
J Gerontol A Biol Sci Med Sci. 2005;
60:660- 666
[129] modrego pj, fayed n, pina ma. Conversion from mild cognitive impairment to probable Alzheimer’s disease predicted by brain magnetic resonance spectroscopy.
Am J Psychiatry. 2005;
162:667- 675
[130] mondadori cr, buchmann a, mustovic h, schmidt cf, boesiger p. Enhanced brain activity may precede the diagnosis of Alzheimer’s disease by 30 years.
Brain. 2006;
129:2908- 2922
[131] mosconi l. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD.
Eur J Nucl Med Mol Imaging. 2005;
32:486- 510
[132] mosconi l, perani d, sorbi s, herholz k, nacmias b. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET.
Neurology. 2004;
63:2332- 2340
[133] mosconi l, tsui wh, de santi s, li j, rusinek h. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis.
Neurology. 2005;
64:1860- 1867
[134] mosconi l, brys m, glodzik-sobanska l, de santi s, rusinek h, de leon mj. Early detection of Alzheimer’s disease using neuroimaging.
Exp. Gerontol. 2007;
42:129- 138
[135] muller mj, greverus d, dellani pr, weibrich c, wille pr. Functional implications of hippocampal volume and diffusivity in mild cognitive impairment.
Neuroimage. 2005;
28:1033- 1042
[136] nagahama y, nabatame h, okina t, yamauchi h, narita m. Cerebral correlates of the progression rate of the cognitive decline in probable Alzheimer’s disease.
Eur Neurol. 2003;
50:1- 9
[137] naggara o, oppenheim c, rieu d, raoux n, rodrigo s. Diffusion tensor imaging in early Alzheimer’s disease.
Psychiatry Res. 2006;
146:243- 249
[138] nestor pj, fryer td, ikeda m, hodges jr. Retrosplenial cortex (BA 29/30) hypometabolism in mild cognitive impairment (prodromal Alzheimer’s disease).
Eur J Neurosci. 2003a;
18:2663- 2667
[139] nestor pj, fryer td, smielewski p, hodges jr. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment.
Ann Neurol. 2003b;
54:343- 351
[140] nestor pj, scheltens p, hodges jr. Advances in the early detection of Alzheimer’s disease.
Nat Med. 2004;
10:S34- S41
[141] norfray jf, provenzale jm. Alzheimer’s disease: neuropathologic findings and recent advances in imaging.
AJR Am J Roentgenol. 2004;
182:3- 13
[142] ohnishi t, matsuda h, tabira t, asada t, uno m. Changes in brain morphology in Alzheimer disease and normal aging: is Alzheimer disease an exaggerated aging process?.
AJNR Am J Neuroradiol. 2001;
22:1680- 1685
[143] olichney jm, morris sk, ochoa c, salmon dp, thal lj. Abnormal verbal event related potentials in mild cognitive impairment and incipient Alzheimer’s disease.
J Neurol Neurosurg Psychiatry. 2002;
73:377- 384
[144] olichney jm, iragui vj, salmon dp, riggins br, morris sk, kutas m. Absent event-related potential (ERP) word repetition effects in mild Alzheimer’s disease.
Clin Neurophysiol. 2006;
117:1319- 1330
[145] pantel j, schonknecht p, essig m, schroder j. Distribution of cerebral atrophy assessed by magnetic resonance imaging reflects patterns of neuropsychological deficits in Alzheimer’s dementia.
Neurosci Lett. 2004;
361:17- 20
[146] pariente j, cole s, henson r, clare l, kennedy a. Alzheimer’s patients engage an alternative network during a memory task.
Ann Neurol. 2005;
58:870- 879
[147] pennanen c, testa c, laakso mp, hallikainen m, helkala el. A voxel based morphometry study on mild cognitive impairment.
J Neurol Neurosurg Psychiatry. 2005;
76:11- 14
[148] perani d, bressi s, cappa sf, vallar g, alberoni m. Evidence of multiple memory systems in the human brain. A [18F]FDG PET metabolic study.
Brain. 1993;
116:903- 919
[149] phillips na, chertkow h, leblanc mm, pim h, murtha s. Functional and anatomical memory indices in patients with or at risk for Alzheimer’s disease.
J Int Neuropsychol Soc. 2004;
10:200- 210
[150] price jc, klunk we, lopresti bj, lu x, hoge ja. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B.
J Cereb Blood Flow Metab. 2005;
25:1528- 1547
[151] prvulovic d, van dv, sack at, maurer k, linden de. Functional activation imaging in aging and dementia.
Psychiatry Res. 2005;
140:97- 113
[152] rauchs g, piolino p, mezenge f, landeau b, lalevee c. Autonoetic consciousness in Alzheimer’s disease: neuropsychological and PET findings using an episodic learning and recognition task.
Neurobiol Aging. 2007, sous presse;
:-
[153] reiman em, caselli rj, yun ls, chen k, bandy d. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E.
N Engl J Med. 1996;
334:752- 758
[154] reiman em, chen k, alexander ge, caselli rj, bandy d. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia.
Proc Natl Acad Sci USA. 2004;
101:284- 289
[155] remy f, mirrashed f, campbell b, richter w. Verbal episodic memory impairment in Alzheimer’s disease: a combined structural and functional MRI study.
Neuroimage. 2005;
25:253- 266
[156] ries ml, schmitz tw, kawahara tn, torgerson bm, trivedi ma, johnson sc. Task-dependent posterior cingulate activation in mild cognitive impairment.
Neuroimage. 2006;
29:485- 492
[157] rombouts sa, barkhof f, veltman dj, machielsen wc, witter mp. Functional MR imaging in Alzheimer’s disease during memory encoding.
AJNR Am J Neuroradiol. 2000;
21:1869- 1875
[158] rosano c, aizenstein hj, cochran jl, saxton ja, de kosky st. Event-related functional magnetic resonance imaging investigation of executive control in very old individuals with mild cognitive impairment.
Biol Psychiatry. 2005;
57:761- 767
[159] rose se, chen f, chalk jb, zelaya fo, strugnell we. Loss of connectivity in Alzheimer’s disease: an evaluation of white matter tract integrity with colour coded MR diffusion tensor imaging.
J Neurol Neurosurg Psychiatry. 2000;
69:528- 530
[160] rose se, mcmahon kl, janke al, o’dowd b, de zubicaray g. Diffusion indices on magnetic resonance imaging and neuropsychological performance in amnestic mild cognitive impairment.
J Neurol Neurosurg Psychiatry. 2006;
77:1122- 1128
[161] sakamoto s, ishii k, sasaki m, hosaka k, mori t. Differences in cerebral metabolic impairment between early and late onset types of Alzheimer’s disease.
J Neurol Sci. 2002;
200:27- 32
[162] salmon e, collette f, degueldre c, lemaire c, franck g. Voxel-based analysis of confounding effects of age and dementia severity on cerebral metabolism in Alzheimer’s disease.
Hum Brain Mapp. 2000;
10:39- 48
[163] sandstrom ck, krishnan s, slavin mj, tran tt, doraiswamy pm, petrella jr. Hippocampal atrophy confounds template-based functional MR imaging measures of hippocampal activation in patients with mild cognitive impairment.
AJNR Am J Neuroradiol. 2006;
27:1622- 1627
[164] saykin aj, flashman la, frutiger sa, johnson sc, mamourian ac. Neuroanatomic substrates of semantic memory impairment in Alzheimer’s disease: patterns of functional MRI activation.
J Int Neuropsychol Soc. 1999;
5:377- 392
[165] scheltens p, launer lj, barkhof f, weinstein hc, jonker c. The diagnostic value of magnetic resonance imaging and technetium 99m-HMPAO single-photon-emission computed tomography for the diagnosis of Alzheimer disease in a community-dwelling elderly population.
Alzheimer Dis Assoc Disord. 1997;
11:63- 70
[166] scheltens p, korf es. Contribution of neuroimaging in the diagnosis of Alzheimer’s disease and other dementias.
Curr Opin Neurol. 2000;
13:391- 396
[167] scheltens p, fox n, barkhof f, de carli c. Structural magnetic resonance imaging in the practical assessment of dementia: beyond exclusion.
Lancet Neurol. 2002;
1:13- 21
[168] schroder j, buchsbaum ms, shihabuddin l, tang c, wei tc. Patterns of cortical activity and memory performance in Alzheimer’s disease.
Biol Psychiatry. 2001;
49:426- 436
[169] shiino a, watanabe t, maeda k, kotani e, akiguchi i, matsuda m. Four subgroups of Alzheimer’s disease based on patterns of atrophy using VBM and a unique pattern for early onset disease.
Neuroimage. 2006;
33:17- 26
[170] shoghi-jadid k, small gw, agdeppa ed, kepe v, ercoli lm. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease.
Am J Geriatr Psychiatry. 2002;
10:24- 35
[171] silverman dh, small gw, chang cy, lu cs, kung de aburto ma. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome.
JAMA. 2001;
286:2120- 2127
[172] small gw. What does imaging add to the management of Alzheimer’s disease?.
CNS Spectr. 2004;
9:20- 23
[173] small gw, kepe v, ercoli lm, siddarth p, bookheimer sy. PET of brain amyloid and tau in mild cognitive impairment.
N Engl J Med. 2006;
355:2652- 2663
[174] sperling ra, bates jf, chua ef, cocchiarella aj, rentz dm. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer’s disease.
J Neurol Neurosurg Psychiatry. 2003;
74:44- 50
[175] starr jm, loeffler b, abousleiman y, simonotto e, marshall i. Episodic and semantic memory tasks activate different brain regions in Alzheimer disease.
Neurology. 2005;
65:266- 269
[176] stern y, moeller jr, anderson ke, luber b, zubin nr. Different brain networks mediate task performance in normal aging and AD: defining compensation.
Neurology. 2000;
55:1291- 1297
[177] takahashi s, yonezawa h, takahashi j, kudo m, inoue t, tohgi h. Selective reduction of diffusion anisotropy in white matter of Alzheimer disease brains measured by 3.0 Tesla magnetic resonance imaging.
Neurosci Lett. 2002;
332:45- 48
[178] teipel sj, willoch f, ishii k, burger k, drzezga a. Resting state glucose utilization and the CERAD cognitive battery in patients with Alzheimer’s disease.
Neurobiol Aging. 2006;
27:681- 690
[179] trivedi ma, schmitz tw, ries ml, torgerson bm, sager ma. Reduced hippocampal activation during episodic encoding in middle-aged individuals at genetic risk of Alzheimer’s disease: a cross-sectional study.
BMC Med. 2006;
4:1
[180] wishart ha, saykin aj, rabin la, santulli rb, flashman la. Increased brain activation during working memory in cognitively intact adults with the APOE epsilon4 allele.
Am J Psychiatry. 2006;
163:1603- 1610
[181] woodard jl, grafton st, votaw jr, green rc, dobraski me, hoffman jm. Compensatory recruitment of neural resources during overt rehearsal of word lists in Alzheimer’s disease.
Neuropsychology. 1998;
12:491- 504
[182] xie s, xiao jx, wang yh, wu hk, gong gl, jiang xx. Evaluation of bilateral cingulum with tractography in patients with Alzheimer’s disease.
Neuroreport. 2005a;
16:1275- 1278
[183] xie s, xiao jx, bai j, jiang xx. Patterns of brain activation in patients with mild Alzheimer’s disease during performance of subtraction: an fMRI study.
Clin Imaging. 2005b;
29:94- 97
[184] xie s, xiao jx, gong gl, zang yf, wang yh. Voxel-based detection of white matter abnormalities in mild Alzheimer disease.
Neurology. 2006;
66:1845- 1849
[185] yetkin fz, rosenberg rn, weiner mf, purdy pd, cullum cm. FMRI of working memory in patients with mild cognitive impairment and probable Alzheimer’s disease.
Eur Radiol. 2006;
16:193- 206
[186] yoshiura t, mihara f, ogomori k, tanaka a, kaneko k, masuda k. Diffusion tensor in posterior cingulate gyrus: correlation with cognitive decline in Alzheimer’s disease.
Neuroreport. 2002;
13:2299- 2302
[187] zahn r, buechert m, overmans j, talazko j, specht k. Mapping of temporal and parietal cortex in progressive nonfluent aphasia and Alzheimer’s disease using chemical shift imaging, voxel-based morphometry and positron emission tomography.
Psychiatry Res. 2005;
140:115- 131
[188] zamrini e, de santi s, tolar m. Imaging is superior to cognitive testing for early diagnosis of Alzheimer’s disease.
Neurobiol Aging. 2004;
25:685- 691
[189] zhang y, schuff n, jahng gh, bayne w, mori s. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease.
Neurology. 2007;
68:13- 19
[190] ziolko sk, weissfeld la, klunk we, mathis ca. Evaluation of voxel-based methods for the statistical analysis of PIB PET amyloid imaging studies in Alzheimer’s disease.
Neuroimage. 2006;
33:94- 102
→ Aller vers SYNTHESE