Effets de xénobiotiques sur la reproduction
2011
15-
Effets et mécanismes d’action
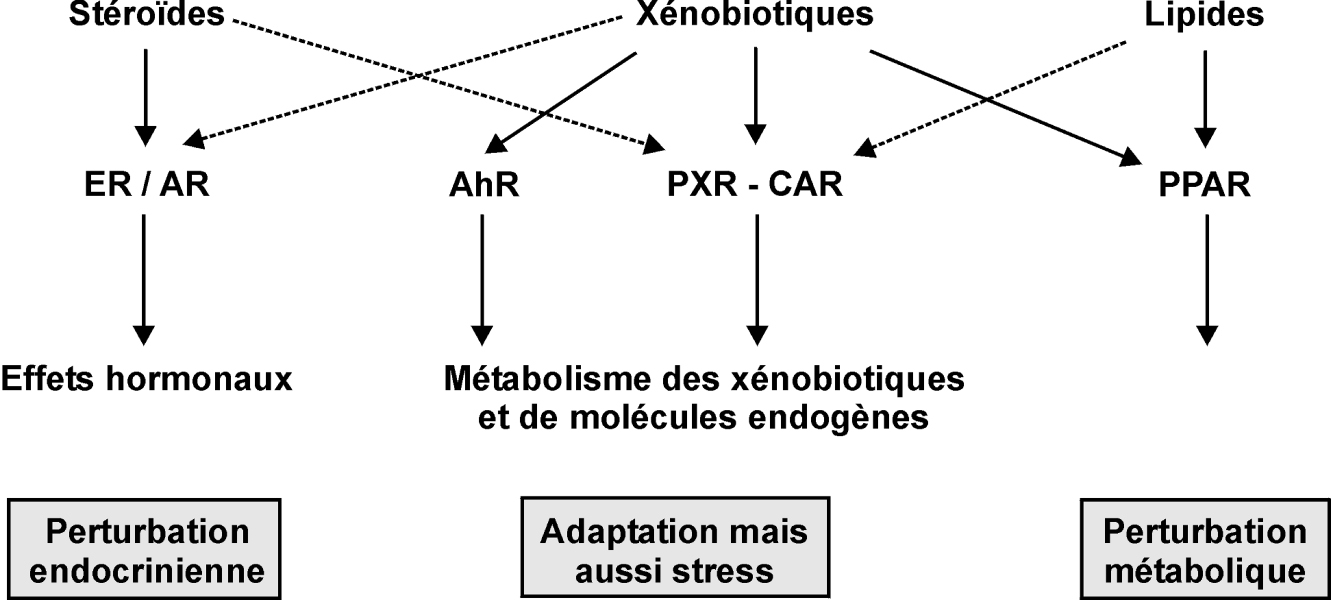
Les différents types d’effets des substances susceptibles d’altérer la fonction de reproduction, peuvent être envisagés en allant de l’échelle la plus fondamentale (perturbation endocrinienne, interaction avec les récepteurs nucléaires) à la plus intégrée (effets sur l’organisme).
Les xénobiotiques peuvent activer différentes catégories de récepteurs que l’on peut classer en deux grands types : les récepteurs des xénobiotiques au sens strict (récepteur AhR de la dioxine et des hydrocarbures aromatiques polycycliques, récepteur PXR capable de lier des médicaments et des pesticides, le récepteur CAR) et les récepteurs de composés endogènes comme les récepteurs hormonaux qui sont susceptibles d’être modulés par ces xénobiotiques (ER α et β, AR). Les récepteurs des xénobiotiques comme PXR ou AhR ont pour fonction principale l’adaptation de l’organisme à l’afflux de xénobiotiques puisqu’ils sont responsables de l’induction de systèmes enzymatiques et de leur élimination. En ce qui concerne les récepteurs à des ligands endogènes, leur activation illégitime par des facteurs environnementaux conduit à une perturbation endocrinienne ou métabolique (figure 15.1

).
La toxicité d’un xénobiotique peut provenir à la fois de l’interaction avec son récepteur légitime et de l’interaction avec des récepteurs de composés endogènes. Une classification schématique des mécanismes d’action est donc difficile, d’autant que l’affinité de ces substances exogènes aux récepteurs de composés endogènes est à prendre en compte : les substances chimiques présentent des affinités souvent très inférieures à celle de l’hormone naturelle.
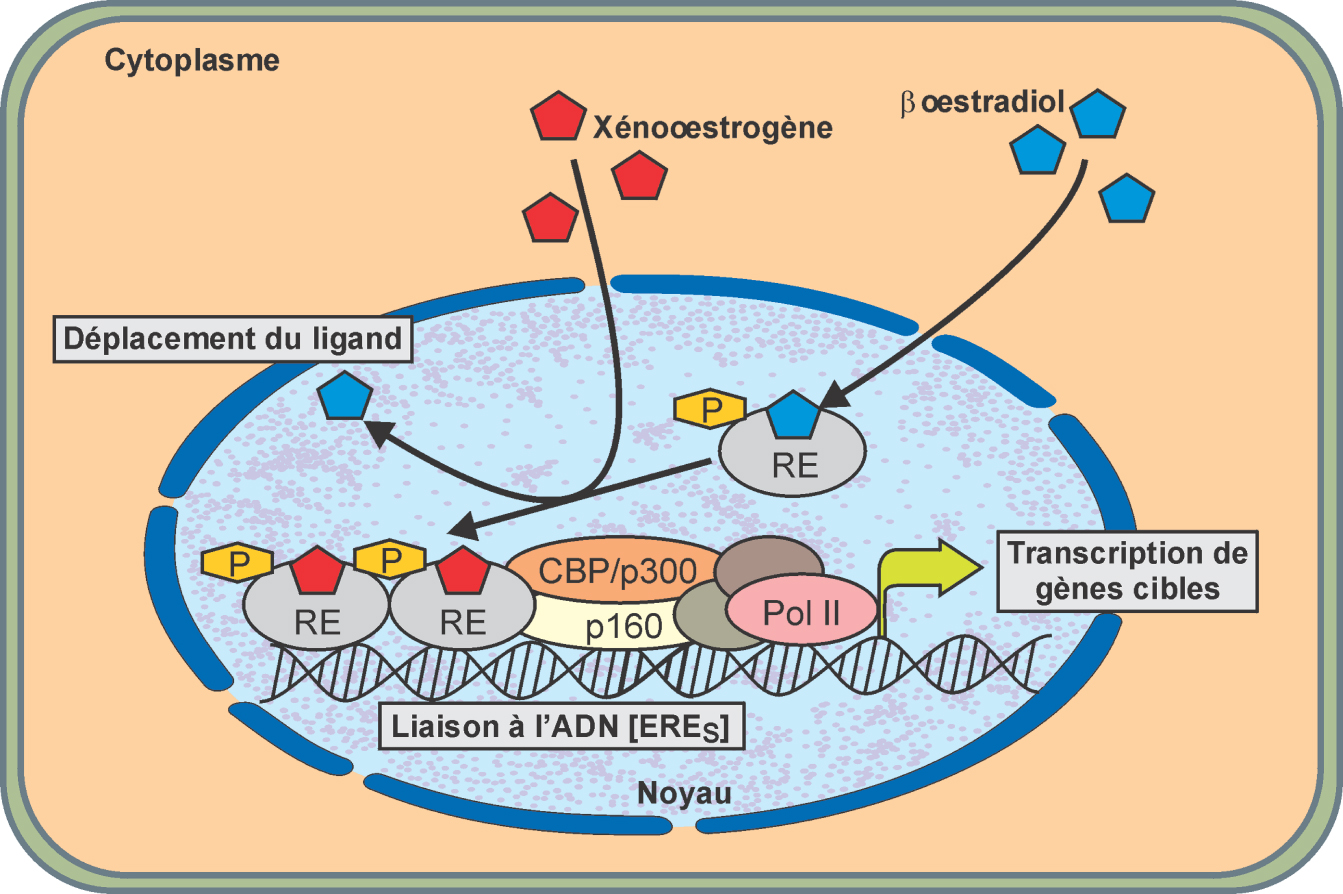
La liaison aux récepteurs nucléaires suivie de l’activation (ou l’inhibition) ne constitue pas le seul mécanisme d’action à l’origine d’un effet toxique. Il est de plus en plus évident que des substances peuvent, comme d’ailleurs certains ligands endogènes des récepteurs nucléaires comme le 17β-estradiol, se fixer sur des récepteurs membranaires. De même, il apparaît de plus en plus que certains récepteurs nucléaires peuvent se localiser sous la membrane plasmique ou dans le cytoplasme et y réguler plusieurs paramètres cellulaires sans pour autant agir sur la transcription de gènes cibles. Si la réalité de ces mécanismes « non-génomiques » est à présent bien établie, ils restent cependant à mieux comprendre, notamment leur détournement possible par des molécules toxiques.
Perturbation endocrinienne
Les perturbateurs endocriniens (PE) peuvent agir à plusieurs niveaux : synthèse des hormones, transport, métabolisme, ou encore liaison avec les récepteurs nucléaires constituant les cibles naturelles des hormones. Leur liaison aux récepteurs nucléaires perturbant la liaison des ligands naturels reste toutefois le mécanisme le plus fréquent.
Les PE sont susceptibles de perturber la synthèse des ligands de récepteurs nucléaires. L’aromatase, enzyme clé de la synthèse de l’œstradiol à partir de la testostérone est une cible bien connue de certains PE.
Ils peuvent altérer la liaison aux protéines de transport comme la SHBG (
sex hormone binding globlulin) ou la transthyrétrine (protéine de transport de la thyroxine). Les PE modifient la clairance des ligands de récepteurs nucléaires en activant des enzymes du métabolisme comme
CYP1A1 (gène cible du récepteur de la dioxine) ou
CYP3A4 et
CYP2B6 (gènes cibles des récepteurs PXR et CAR). Ils peuvent influencer la concentration en récepteurs nucléaires. Les œstrogènes contrôlent l’expression de certains récepteurs nucléaires comme PR (Kastner et coll., 1990
) et RAR α (Laganiere et coll., 2005
) et la perturbation œstrogénique peut donc par ricochet agir sur d’autres systèmes hormonaux. Les ligands de AhR en activant leur récepteur, lui permettent de devenir nucléaire, de s’associer aux récepteurs ER et AR et d’accélérer leur dégradation par le protéasome (Ohtake et coll., 2007
).
Enfin, les PE régulent l’expression de gènes importants pour le développement du tractus reproducteur et participent aux modifications épigénétiques du génome.
Familles des récepteurs nucléaires
Les récepteurs nucléaires (codés par 48 gènes distincts chez l’homme) sont regroupés en plusieurs sous-familles qui incluent les récepteurs aux hormones thyroïdiennes (TR), à l’acide rétinoïque (RAR), à la vitamine D (VDR), à l’ecdysone (EcR), les récepteurs contrôlant l’activation de la prolifération des péroxysomes (PPAR), ainsi que les récepteurs des xénobiotiques comme PXR ou CAR, les récepteurs aux rétinoïdes (RXR), les récepteurs aux hormones stéroïdiennes telles que les glucocorticoïdes (GR), les androgènes (AR), les minéralocorticoïdes (MR), les progestatifs (PR, pour
Progesterone Receptor), les œstrogènes (ER), ainsi que les récepteurs orphelins comme ERR (
Estrogen-Related Receptor), SHP (
Short Heterodimer Partner) (Escriva et coll., 2000
; Aranda et coll., 2001
). Ces récepteurs nucléaires (RN) sont classés en six groupes selon une nomenclature phylogénique de forme NRXYZ où X représente la sous-famille, Y indique le groupe et Z, le gène (
Nuclear Receptors Nomenclature Committee 1999, tableau 15.I
).
Par exemple, les récepteurs des œstrogènes α et β, qui appartiennent respectivement à la catégorie NR3A1 et NR3A2, sont classés dans la troisième famille des RN, aux côtés des ERR, GR (Glucocorticoid Receptor), MR (Mineralocorticoid Receptor), PR et AR (Androgen Receptor). Les ER forment alors le premier groupe (A) de cette sous-famille et sont codés par deux gènes, désignés respectivement 1 et 2 ce qui correspond aux formes ERα et ERβ. Le groupe B est composé par les ERR et les autres récepteurs nucléaires de cette troisième famille appartiennent au groupe C.
Un grand nombre de récepteurs dont les ligands naturels ne sont pas connus sont appelés récepteurs nucléaires orphelins et ont été identifiés par leur similarité de séquence avec des facteurs existants. Parmi ces récepteurs, nous retrouvons les ERR présents dans la classe III des RN.
Les récepteurs nucléaires sont au cœur de la régulation de nombreux réseaux de gènes impliqués dans la prolifération et la différenciation cellulaire, le développement, l’homéostasie et le métabolisme (Gronemeyer et coll., 2004
). Leur dysfonctionnement, mutation ou (in)activation inappropriée, peut conduire à des pathologies de la reproduction ou du métabolisme comme les cancers hormono-dépendants, la stérilité, le diabète ou l’obésité (Swedenborg et coll., 2009
). Les récepteurs nucléaires sont des protéines modulaires composées de plusieurs domaines dont un domaine de liaison au ligand qui joue un rôle important dans leur activation. De nombreux perturbateurs endocriniens peuvent se lier à des récepteurs nucléaires et conduire à leur activation ou inactivation.
Tableau 15.I Récepteurs nucléaires humains (d’après Gronemeyer et coll., 2004)
|
Nom
|
Abréviation
|
Nomenclature
|
Ligand
|
|
Récepteur de l’hormone thyroïdienne
|
TRα
|
NR1A1
|
Hormone thyroïdienne
|
|
TRβ
|
NR1A2
|
Hormone thyroïdienne
|
|
Récepteur de l’acide rétinoïque
|
RARα
|
NR1B1
|
Acide rétinoïque
|
|
RARβ
|
NR1B2
|
Acide rétinoïque
|
|
RARγ
|
NR1B3
|
Acide rétinoïque
|
|
Récepteur de prolifération des peroxysomes (Peroxisome proliferator-activated receptor)
|
PPARα
|
NR1C1
|
Acides gras, leukotriène B4, fibrates
|
|
PPARβ
|
NR1C2
|
Acides gras
|
|
PPARγ
|
NR1C3
|
Acides gras, prostaglandine J2
|
|
Reverse erbA
|
Rev-erbα
|
NR1D1
|
Orphelin
|
|
Rev-erbβ
|
NR1D1
|
Orphelin
|
|
RAR-related orphan receptor
|
RORα
|
NR1F1
|
Cholestérol, cholestéryl sulfate
|
|
RORβ
|
NR1F2
|
Acide rétinoïque
|
|
RORγ
|
NR1F3
|
Acide rétinoïque
|
|
Récepteur hépatique X
|
LXRα
|
NR1H3
|
Oxystérols, T090 1317, GW3965
|
|
LXRβ
|
NR1H2
|
Oxystérols, T090 1317, GW3965
|
|
Récepteur Farnesoïde X
|
FXRα
|
NR1H4
|
Acides biliaires, fexaramine
|
|
FXRβa
|
NR1H5
|
Lanostérol
|
|
Récepteur de la vitamine D
|
VDR
|
NR1I1
|
1,25-dihydroxy vitamine D3, acide litocholique
|
|
Récepteur prégnane X
|
PXR
|
NR1I2
|
Xénobiotiques, PCNb
|
|
Récepteur constitutif de l’androstane
|
CAR
|
NR1I3
|
Xénobiotiques, phénobarbital
|
|
Facteur nucléaire humain 4
|
HNF4α
|
NR2A1
|
Orphelin
|
|
HNF4β
|
NR2A2
|
Orphelin
|
|
Récepteur X de l’acide rétinoïque
|
RXRα
|
NR2B1
|
Acide rétinoïque
|
|
RXRβ
|
NR2B2
|
Acide rétinoïque
|
|
RXRγ
|
NR2B3
|
Acide rétinoïque
|
|
Récepteur testiculaire
|
TR2
|
NR2C1
|
Orphelin
|
|
TR4
|
NR2C2
|
Orphelin
|
|
Tailless
|
TLL
|
NR2E2
|
Orphelin
|
|
Récepteur nucléaire spécifique des cellules photorécepteurs
|
PNR
|
NR2E3
|
Orphelin
|
|
Facteur de transcription du promoteur de l’ovalbumine de poulet
|
COUP-TFI
|
NR2F1
|
Orphelin
|
|
COUP-TFII
|
NR2F2
|
Orphelin
|
|
ErbA2-related gene-2
|
EAR2
|
NR2F6
|
Orphelin
|
|
Récepteur des œstrogènes
|
ERα
|
NR3A1
|
17β-Œstradiol, Tamoxifène, Raloxifène
|
|
ERβ
|
NR3A2
|
17β-Œstradiol, plusieurs composés synthétiques
|
|
Récepteur « like » des œstrogènes
|
ERRα
|
NR3B1
|
Orphelin
|
|
ERRβ
|
NR3B2
|
DESc 4-OH tamoxifène
|
|
ERRγ
|
NR3B3
|
DES, 4-OH tamoxifène
|
|
Récepteur des glucocorticoïdes
|
GR
|
NR3C1
|
Cortisol, dexaméthasone, RU486
|
|
Récepteur des minéralocorticoïdes
|
MR
|
NR3C2
|
Aldostérone, spironolactone
|
|
Récepteur de la progestérone
|
PR
|
NR3C3
|
Progestérone, acétate de médroxyprogestérone, RU486
|
|
Récepteur des androgènes
|
AR
|
NR3C4
|
Testostérone, flutamide
|
|
Facteur B induit par le NGFd
|
NGFIB
|
NR4A1
|
Orphelin
|
|
Nur related factor 1
|
NURR1
|
NR4A2
|
Orphelin
|
|
Neuron-derived orphan receptor 1
|
NOR1
|
NR4A3
|
Orphelin
|
|
Facteur stéroïdogénique 1
|
SF1
|
NR5A1
|
Orphelin
|
|
Liver receptor homologous protein 1
|
LRH1
|
NR5A2
|
Orphelin
|
|
Récepteur des cellules germinales
|
GCNF
|
NR6A1
|
Orphelin
|
|
DSS-AHCe critical region on the chromosome, gene 1
|
DAX1
|
NR0B1
|
Orphelin
|
|
Short heterodimeric partner
|
SHP
|
NR0B2
|
Orphelin
|
a XRβ est un pseudogène et ne code pas de récepteur fonctionnel ; b PCN : pregnenolone 16α-carbonitrile ; c DES : diéthylstilbestrol ; d NGF : nerve growth factor ; e DSS-AHC : dosage-sensitive sex reversal-adrenal hypoplasia congenita
Mécanisme général d’action des récepteurs nucléaires
Comme leur nom l’indique, la plupart des récepteurs nucléaires sont présents dans le noyau cellulaire où ils régulent la transcription des gènes. Il existe certaines exceptions comme le récepteur des glucocorticoïdes ou celui des androgènes qui sont localisés dans le cytoplasme en absence de ligand, la liaison à celui-ci induisant la translocation du récepteur au noyau. En outre, des études récentes suggèrent que certains récepteurs même en présence de leur ligand peuvent agir dans le cytoplasme d’une façon non génomique en interagissant avec des cibles dont l’identité reste encore peu claire.
De nombreuses données fonctionnelles indiquent qu’en absence de leur ligand la plupart des récepteurs nucléaires sont fixés à l’ADN, reconnaissent des corépresseurs transcriptionnels et bloquent la transcription. C’est le cas par exemple de PPAR qui forme un hétérodimère avec RXR et qui réprime la transcription en absence de ligand. Cependant, plusieurs analyses pangénomiques récentes fondées sur la méthode d’immunoprécipitation de chromatine couplée à du séquençage massif remettent quelque peu en cause la généralité de ce mécanisme et suggèrent que cela ne se produit pas pour tous les gènes cibles. Par ailleurs, il faut noter que ce mécanisme ne s’applique pas aux récepteurs stéroïdiens comme ER, AR ou GR qui ne semblent pas présents dans le noyau en absence de ligand.
Il est clairement établi que le ligand induit un changement de conformation du domaine de fixation du ligand (LBD) via la modification de la surface du récepteur, à l’origine du départ du corépresseur et du recrutement du coactivateur. C’est le cas des récepteurs qui hétérodimérisent avec RXR comme les récepteurs des rétinoïdes (RAR α, β et γ), de la vitamine D (VDR), des xénobiotiques (PXR, CAR), des TR et des PPAR. Ces récepteurs ont des activités basales qui varient selon leur affinité pour les corépresseurs. Ainsi les TR sont de meilleurs répresseurs que PXR ou CAR. De même, PPARβ est plus répresseur que PPARγ (Shi et coll., 2002
).
Concept de Selective Nuclear Receptor Modulators (SnuRM)
L’étude détaillée de la pharmacologie des récepteurs nucléaires a permis de mieux comprendre les bases structurales et fonctionnelles de leur régulation par des ligands agonistes ou antagonistes. Ceci a été rendu possible par l’intérêt clinique que pourraient présenter des molécules capables d’activer un récepteur donné seulement dans certains organes de façon à avoir une action dénuée le plus possible d’effets secondaires. Ce concept est bien illustré par le cas des récepteurs des œstrogènes qui ont été sans doute les plus étudiés de ce point de vue mais on sait à présent qu’il est généralisable à l’ensemble des récepteurs nucléaires (Gronemeyer et coll., 2004
; Germain et coll., 2006
). Ainsi il serait important de pouvoir développer des molécules capables d’activer le récepteur ERα dans l’os (pour traiter les conséquences de l’ostéoporose suite à la chute du taux d’œstrogènes qui se produit chez la femme lors de la ménopause) tout en l’inactivant dans l’ovaire, l’utérus ou le sein pour éviter les risques de cancers associés à la stimulation œstrogénique. Une telle molécule, le raloxifène a été mise au point et son mode de fonctionnement tissu-spécifique repose sur deux principes importants du fonctionnement des récepteurs nucléaires. Selon le premier principe, différents ligands en se fixant dans la poche hydrophobe du domaine de fixation du ligand (LBD) induisent un changement de conformation subtilement différent. Ce changement de conformation aboutit à la rélocalisation d’une hélice α C-terminale du LBD, l’hélice 12 qui va former une surface du récepteur disponible pour l’interaction avec des coactivateurs transcriptionnels. Il a été montré que les positions de l’hélice 12 dans des LBD de ERα fixant le 17β-œstradiol, un antagoniste comme le tamoxifene ou la génisteine sont effectivement différentes.
Selon le deuxième principe, il existe de nombreux coactivateurs transcriptionels des récepteurs nucléaires, chacun d’entre eux ayant une sensibilité différente à des changements de la surface du récepteur induits par des ligands différents (Shang et Brown, 2002
). Ces coactivateurs ayant des distributions tissulaires différentes, on arrive ainsi à comprendre comment un ligand donné peut activer un récepteur dans un tissu A donné mais pas dans un autre tissu B. Dans A, le récepteur lié au ligand aura une conformation reconnue par un coactivateur présent dans ce tissu alors que dans le tissu B ce coactivateur ne sera pas présent et donc le récepteur sera inactif. Ceci débouche sur le concept de SERM (
selective estrogen receptor modulator) : des ligands modulateurs du récepteur des œstrogènes qui peuvent être sélectifs d’un tissu donné.
Ce concept est bien établi et il a pu être appliqué à de nombreux autres récepteurs (AR, PPAR, TR...) au point d’être à présent généralisé à l’ensemble de la superfamille. On parle donc de SnuRM : Selective Nuclear Receptor Modulators. Cependant, la transcription est un processus très dynamique et ceci n’est pas pris en compte dans le modèle décrit. Ce concept reste néanmoins fondamentalement important pour l’étude des molécules toxiques et en particulier des perturbateurs endocriniens. En effet, ces molécules semblent pouvoir être considérées comme des SNuRM ou SERM. Chacune d’entre elles peut induire une conformation légèrement différente du récepteur qu’elle va pouvoir fixer et entraîner un panel d’effets dans différents organes spécifiques de chaque molécule. Ceci est d’autant plus vrai que, la plupart des perturbateurs endocriniens n’ont pas une cible unique mais sont capables de reconnaître plusieurs récepteurs avec des affinités différentes. La gamme des effets générés par des molécules différentes est donc très vaste et cela explique pourquoi il est très compliqué de pouvoir faire un lien entre les mécanismes d’action d’une molécule in vitro et ses effets in vivo. Ce n’est pas parce que deux molécules vont in vitro être capables de se fixer au même récepteur avec des affinités comparables qu’elles vont avoir in vivo une gamme d’effets identiques, même si leur toxicocinétique, et leur métabolisme sont très proches.
Récepteurs ER
Les récepteurs des œstrogènes (ERα et β, NR3A1 et NR3A2) sont les récepteurs de l’hormone sexuelle féminine, l’œstradiol qui joue un rôle très important dans une grande variété de tissus comme la glande mammaire, l’utérus, la moelle osseuse, l’os et le système cardiovasculaire. ERα est surtout exprimé dans l’utérus, le foie, les reins et le cœur alors que ERβ est plutôt exprimé dans l’ovaire, la prostate, les poumons, le tractus intestinal et les systèmes hématopoïétique et nerveux central (Kuiper et coll., 1997
). Ils sont également co-exprimés dans un certain nombre de tissus comme la glande mammaire, la thyroïde, la moelle osseuse et certaines régions du cerveau. Quoique ayant des mécanismes similaires d’action, des différences existent dans leur pharmacologie et dans leur capacité à activer les gènes cibles ce qui suggère que ces récepteurs ont des rôles différents comme l’a démontré l’étude des souris dans lesquelles les gènes codant pour chacun de ces récepteurs ont été inactivés (Couse et coll., 1997
). Ainsi quand les deux récepteurs sont co-exprimés, ERβ possède une action inhibitrice sur la capacité de ERα à activer ses gènes cibles. De même, ERβ a été montré comme capable de bloquer l’effet activateur de ERα sur la prolifération dans le sein, l’utérus ou la prostate (Pettersson et coll., 2000
).
La grande majorité des ligands environnementaux de ces récepteurs sont des molécules agonistes et ont une meilleure affinité pour ERα (figure 15.2
). Cependant, certaines molécules ont une meilleure affinité pour ERβ. C’est le cas de phyto-œstrogènes comme la génistéine (chez les mammifères), la biochanine A ou la daidzéine, de benzophénones comme la benzophénone 1 ou 2, de biphénol comme le 44’ biphénol. Enfin, certaines molécules ont une activité agoniste sur ERα et antagoniste sur ERβ. C’est le cas de pesticides comme le méthoxychlore ou le chlordécone (Le Maire et coll., 2010
). Ceci illustre bien le fait que de nombreuses molécules toxiques se comportent comme des SERM et peuvent avoir des effets tissus-spécifiques marqués ce qui rend la comparaison
in vitro in vivo très complexe.
Récepteurs AR
Le récepteur des androgènes (AR, NR3C4) est le récepteur des hormones sexuelles mâles, la testostérone et son métabolite, la dihydrotestostérone. L’AR est principalement exprimé dans le testicule, il est présent dans la prostate, les glandes surrénales, les reins, le cerveau et l’hypophyse. Le rôle du récepteur AR dans les organes mâles est très similaire à celui des récepteurs ER dans les organes femelles. Les androgènes humains comme la testostérone ou la dihydrotestostérone sont des agonistes. Chez les poissons, le système de réponse aux androgènes est plus complexe : il existe un second ligand en plus de la dihydrotestostérone, la 11-ketotestostérone (11KT) et la plupart des espèces présentent deux récepteurs des androgènes AR-A et AR-B dont les pharmacologies semblent être distinctes (Douard et coll., 2008
). La grande majorité des ligands environnementaux de ce récepteur sont des molécules antagonistes (Paris et coll., 2002
; Korner et coll., 2004
). Très peu d’androgènes environnementaux sont des agonistes excepté le retardateur de flamme bromé TBECH (Khalaf et coll., 2009
).
Récepteurs PPAR
La famille des récepteurs activés par les inducteurs de la prolifération des péroxysomes (PPAR) comprend trois membres distincts, désignés α, β et γ (NR1C1, NR1C2, NR1C3). Ils sont activés par la liaison de certains acides gras et/ou de leurs métabolites lipidiques (Forman et coll., 1997
; Kliewer et coll., 1997
). Les PPAR pourraient ainsi jouer un rôle déterminant en signalant, au niveau de l’expression génique, un changement de l’apport nutritionnel et, en particulier, de sa composition lipidique. Les PPAR forment des hétérodimères avec les récepteurs RXR (Gronemeyer et coll., 2004
) et se fixent sur des éléments de réponse qui sont des répétitions directes du motif hexamérique de reconnaissance des récepteurs nucléaires espacés par un nucléotide.
Les PPAR présentent une expression tissulaire spécifique. Chez l’homme, PPARα est la forme majoritaire dans le foie et est exprimé à un niveau relativement faible dans les autres tissus (Auboeuf et coll., 1997
). PPARβ est exprimé de façon très ubiquitaire avec, peut-être, une expression plus importante dans le côlon et dans la peau. Le même type de résultat a été retrouvé chez le rat adulte, chez qui PPARγ est exprimé principalement dans le tissu adipeux et le tractus gastro-intestinal, en particulier dans le côlon, alors qu’il est très faiblement représenté dans le foie ou le muscle squelettique (Auboeuf et coll., 1997
).
Les ligands environnementaux de PPARγ sont essentiellement agonistes. Ce sont certains phtalates (MEHP, BBP, DBP), les PFOS et PFOA et des dérivés halogénés du bisphénol A (Casal-Casas et coll., 2008
; Balaguer, communication personnelle). Les organoétains activent l’hétérodimère RXRα-PPARγ mais l’activation pourrait se produire essentiellement à travers RXRα (Le Maire et coll., 2009
).
Chez les rongeurs, une spécificité des PPAR doit être mentionnée. En effet, ceux-ci sont appelés PPAR pour
Peroxysome Proliferators Activated Receptors parce que, lors de leur découverte, on a pu montrer qu’ils étaient activés par des hépatocarcinogènes comme la nafénopine (Issemann et Green, 1990
), qui sont connus pour promouvoir une prolifération des peroxysomes liée à leur activité carcinogénique. Depuis, il a été démontré que cette prolifération des peroxysomes ne se produit que chez les rongeurs et n’est pas présente dans d’autres espèces y compris l’homme. Par ailleurs, il est intéressant de noter que le MEHP a été démontré être un activateur des PPAR dès la découverte de ces derniers (Issemann et Green, 1990
).
Récepteurs des hormones thyroïdiennes
Les hormones thyroïdiennes interviennent dans le développement des vertébrés notamment la métamorphose chez les amphibiens et les poissons et le maintien de l’homéostasie métabolique. Leurs actions sont médiées par les récepteurs nucléaires TRα et TRβ (NR1A1, NR1A2). La différence d’expression tissulaire de ces deux récepteurs suggère qu’ils exercent des fonctions physiologiques non redondantes. TRβ semble principalement impliqué dans le rétrocontrôle négatif de la sécrétion hypophysaire de TSH (thyrotropine), dans la physiopathologie de la résistance aux hormones thyroïdiennes (RHT), et dans le développement de l’audition.
Au contraire, les souris dont le gène de TRα été invalidé présentent une atrophie thyroïdienne avec diminution des taux de TSH et d’hormones thyroïdiennes. Le phénotype clinique associe un retard de la croissance et de la maturation osseuse et intestinale et l’absence de déficit auditif. Cependant les récepteurs TRα et TRβ sont capables, dans une certaine mesure, de coopérer et/ou de se substituer l’un à l’autre (Flamant et coll., 2007
). Certains organes constituent cependant des tissus cibles spécifiques d’un type de récepteur comme l’oreille interne, l’hypophyse, le cœur, le foie, l’os ou l’intestin grêle.
Le bisphénol A et ses dérivés halogénés ont été décrits comme étant des antagonistes des TR (Moriyama et coll., 2002
; Sun et coll., 2009
).
Régulation de l’expression de gènes importants pour le développement du tractus reproducteur
Il existe deux grands exemples bien caractérisés de la régulation de gènes impliqués dans le développement du tractus reproducteur. Tout d’abord, l’analyse du promoteur du gène de l’INSL3 (
Insuline-like 3) a permis de montrer que celui-ci contenait à la fois des éléments de réponse aux androgènes et aux œstrogènes ce qui en fait donc une cible de choix pour l’étude des perturbateurs endocriniens chez le mâle (Laguë et Tremblay, 2009
). La testostérone stimule l’expression de ce gène et les œstrogènes l’inhibent. Chez la femelle, le gène à homeobox
HOXA10 est critique pour l’organogenèse utérine. L’expression de celui-ci est dépendante de la présence d’un élément de réponse aux œstrogène (ERE) et peut être altérée par ER α (Akbas et coll., 2004
; Couse et coll., 2004
). Le DES est capable d’altérer l’expression de ce gène, conduisant à son expression de manière inappropriée, et d’induire ainsi des malformations du tractus génital femelle. Plus récemment, il a également été proposé que le DES augmente la méthylation de ce gène spécifiquement lors d’une exposition
in utero (Bromer et coll., 2009
).
Bien que beaucoup moins clairement caractérisée, l’altération des enzymes de biosynthèse des stéroïdes a été rapportée en réponse à divers PE. Ainsi par exemple les phtalates inhibent l’expression des enzymes de biosynthèse de la testostérone dans le testicule fœtal de rat tels que Star, Cyp11a et Cyp17a. Dans les cellules de la granulosa, le bisphénol A pourrait altérer l’expression de l’aromatase, enzyme impliquée dans la transformation de la testostérone en œstradiol (Kwintkiewicz et coll., 2010
). Cependant, il n’est pas évident que ces mécanismes impliquent des récepteurs nucléaires. L’inhibition directe de l’activité de ces enzymes est également proposée comme un mécanisme d’action pour certains perturbateurs endocriniens.
L’application actuelle des approches de génomique (microarray et séquençage haut débit) et de protéomique apparaît comme une possibilité pour prochainement identifier globalement l’ensemble des gènes régulés par un perturbateur endocrinien donné. Cependant, à l’heure actuelle les travaux utilisant ces technologies dégagent essentiellement des « signatures » de perturbateurs sans apporter encore un lien de causalité entre la variation d’expression des gènes identifiés et la survenue de pathologies.
Plusieurs études ont montré que des perturbateurs endocriniens peuvent induire des signatures différentes de celles des ligands endogènes, notamment les œstrogènes. Cela renforce la notion que ces perturbateurs ont une vaste gamme d’effets qu’il faut étudier en tenant compte de leur spécificité d’action et de la multiplicité des cibles possibles. L’idée d’un lien simple entre perturbation endocrinienne, action œstrogénique et reprotoxicité doit être abandonnée, la situation apparaissant bien plus complexe.
Mécanisme épigénétique
L’épigénèse est un mécanisme d’altération du génome sans modification de la séquence d’ADN, basé sur un changement dans la méthylation de l’ADN et de l’acétylation des histones de la chromatine. Ces modifications perdurent sur plusieurs générations et peuvent donc provoquer un changement de phénotype sous l’influence de l’environnement. Cependant, il n’est pas toujours évident de distinguer un réel mécanisme épigénétique d’un effet génotoxique ou d’un dommage oxydatif. Le stress oxydant impliqué dans un grand nombre de mécanismes physiopathologiques, est un effet secondaire fréquent de l’exposition à des polluants, en particulier par l’intermédiaire de l’induction de cytochromes P450. Des polluants peuvent entraîner également l’induction de cytokines et une situation inflammatoire qui peut être liée au stress oxydant.
Effets épigénétiques dans la lignée germinale
Une des spécificités de la lignée germinale est la transmission non seulement du génome mais également d’une mémoire épigénétique à la génération suivante. Il a été montré récemment qu’une exposition transitoire au cours de l’organogenèse gonadique à deux perturbateurs endocriniens, la vinclozoline ou le méthoxychlore, réduit la fertilité et la production de sperme du testicule adulte (Anway et coll., 2005
). De manière frappante ce phénotype est transmis à travers la lignée germinale mâle sur au moins quatre générations sans exposition additionnelle. Ce phénotype a été associé à une modification globale de la méthylation du génome dans la lignée germinale mâle. Ce phénomène a ouvert un nouveau champ de recherche au travers des effets épigénétiques induits par les perturbateurs et qui pourraient être transmis de génération en génération. Notons immédiatement deux points. La fenêtre d’exposition proposée correspond à une étape du développement des cellules germinales primordiales au cours de laquelle la méthylation de l’ADN subit de profonds bouleversements, étant déméthylée puis reméthylée de manière sexe-spécifique. Les doses utilisées de substances sont cependant très nettement supérieures à celles pouvant être retrouvées dans l’environnement.
Effets au niveau cellulaire
Au sein des organes composant le tractus reproducteur, de nombreux paramètres cellulaires sont mesurés : apoptose, prolifération, différenciation. La perturbation de ces processus peut être source de troubles de la fertilité ou expliquer la survenue de lésions précancéreuses. Comme indiqué ci-dessus, établir un lien entre l’action in vitro des différentes substances, les effets cellulaires observés lors d’une exposition expérimentale et les anomalies physiopathologiques éventuellement observées chez l’homme ou l’animal est extrêmement difficile.
Apoptose ou mort cellulaire programmée
C’est une voie de mort cellulaire en réponse à un signal et qui aboutit à la fragmentation de l’ADN ; elle est physiologique et programmée. L’apoptose est en équilibre constant avec la prolifération cellulaire. Dans le cas du développement des gonades, plusieurs perturbateurs endocriniens sont suspectés d’augmenter l’apoptose au sein de la lignée germinale. L’apoptose des cellules germinales conduit à une diminution du nombre des cellules germinales, un processus qui, s’il n’est pas compensé par la prolifération des cellules survivantes, peut conduire à une diminution du nombre de gamètes et donc à une hypofertilité. Ainsi par exemple des stress génotoxiques tels que les rayonnements ionisants sont connus depuis longtemps pour induire l’apoptose des cellules germinales tant mâles que femelles et peuvent ainsi être cause de stérilité. Plusieurs phtalates ou le DES (diéthylstilbestrol) ont été décrits comme pouvant induire l’apoptose des cellules germinales mâles au cours du développement (Habert et coll., 2009
). Récemment, la génisteïne a été montrée capable d’induire de l’apoptose
in vivo chez le zebrafish traité au cours de la période embryonnaire (Sassi-Mesai et coll., 2009
).
L’apoptose est un phénomène physiologiquement impliqué dans le développement des organes et une baisse de l’apoptose peut également avoir des conséquences pathologiques. Les cellules cancéreuses sont fréquemment des cellules dans lesquelles ce mécanisme d’apoptose fonctionne mal et survivent en dépit d’anomalies qui auraient dû conduire à leur élimination. Ainsi, l’apparition ou le développement de lésions « précancéreuses » est souvent attribuée à un défaut dans une voie d’apoptose. Le gène
P53, un acteur majeur de l’apoptose, est un gène dit suppresseur de tumeur. Dans le cadre des perturbateurs endocriniens, notons par exemple qu’une exposition périnatale au bisphénol A peut diminuer l’apoptose dans la glande mammaire des souris pubères (Munoz de Toro, 2005
).
Par ailleurs, l’effet sur l’apoptose peut varier selon l’âge ou le stade de développement. Chez la souris, il a été montré que le MEHP (mono-éthylhexyl phtalate) induit l’apoptose des cellules germinales fœtales mâles à 13,5 jours post-conception mais a très peu d’effet à dose équivalente à 15,5 jours post-conception et est à nouveau capable d’induire l’apoptose des cellules germinales dans des testicules de 18,5 jours post-conception (Lehraiki et coll., 2009
). De telles fenêtres d’action extrêmement spécifiques sont couramment décrites pour la gamétogenèse et la stéroïdogenèse fœtale, deux fonctions dont la mise en place est extrêmement dynamique.
Prolifération cellulaire
Un dérèglement de la prolifération cellulaire peut également induire des troubles de la fertilité ou être suspecté dans la survenue de cancers. Notons d’ailleurs que de nombreux gènes contrôlant le cycle cellulaire sont également appelés « suppresseur de tumeurs ».
La diminution de l’activité prolifératrice d’un type cellulaire peut être due à la surexpression d’inhibiteur du cycle cellulaire tel que les protéines p16, p21 ou p27 qui inhibent les complexes Cdk/cyclines requis pour la progression à travers les différentes phases du cycle cellulaire. À titre d’exemple, les hormones thyroïdiennes inhibent la prolifération des cellules de Sertoli en augmentant l’expression de p27 dans le testicule postnatal et la conséquence de ceci est une diminution du poids testiculaire à l’âge adulte ainsi que de la réserve spermatique (nombre de spermatozoïdes) (Holsberger et coll., 2005
; Holsberger et Cooke, 2005
). Dans le cadre des perturbateurs endocriniens, citons l’exemple des phtalates (DBP) qui sont eux aussi capables de diminuer la prolifération des cellules de Sertoli chez le rat (Auharek et coll., 2010
).
Différenciation cellulaire
Dans les tissus, des cellules souches, multipotentes ou progénitrices, se différencient ; la perturbation de ces processus de différenciation peut être cause de troubles de la fertilité ou de cancer.
Dans la lignée germinale, les cellules mitotiques (cellules germinales primordiales, gonocytes, ovogonies ou spermatogonies) expriment de nombreux marqueurs de cellules souches tels que OCT4, un facteur de transcription. Au moment de la différenciation de ces cellules, celles-ci perdent leurs marqueurs de multipotence. Le blocage de la différenciation de cellules germinales fœtales est corrélé à la survenue de tumeurs testiculaires. Ainsi l’invalidation du gène
Dmrt1 chez la souris induit le maintien de l’expression de Oct4, Sox2 et Nanog dans les cellules germinales dans le testicule fœtal et la survenue de tératome dans le testicule des souris adultes (fond génétique SV129, Krentz et coll., 2009
). Ainsi, pour les perturbateurs endocriniens, il a été proposé que certains phtalates (DBP) bloquent ou retardent la différenciation des cellules germinales fœtales mâles chez le rat en maintenant l’expression d’OCT4 dans de petits groupes de cellules qui auraient échappé au processus de différenciation (Ferrara et coll., 2006
). Cependant, il n’a pas été retrouvé de cancers testiculaires chez les rats exposés aux phtalates peut-être du fait que ce type de cancer est très peu fréquent chez les rongeurs en dehors de certains fonds génétiques très spécifiques.
Dans le cas des tissus stéroïdogéniques, cellules de la granulosa ou cellules de Leydig, la sécrétion d’hormones stéroïdes en réponse à une stimulation par des gonadotropines est couramment utilisée comme un paramètre permettant de juger de l’état de différenciation. Ainsi, la sécrétion de testostérone par les cellules de Leydig en réponse à la LH est un paramètre permettant de mesurer la différenciation des cellules de Leydig (Livera et coll., 2006
). Dans le cas des cellules de la granulosa, c’est la sécrétion de progestérone et/ou d’œstradiol qui est mesurée. Par exemple, il a été montré que le bisphénol A diminue la production de progestérone des cellules de la granulosa porcines (Grasselli et coll., 2010
).
Interaction avec plusieurs voies de signalisation
Le récent travail de David Volle qui s’est intéressé aux effets du DES (diéthyl stilbestrol, un œstrogène synthétique non stéroïdien) dans le testicule postnatal de souris, illustre bien ce phénomène d’interaction avec plusieurs voies de signalisation (Volle et coll., 2009
). En effet, cette équipe a montré que les perturbations des fonctions testiculaires induites par l’injection de DES à des souriceaux impliquent à la fois une perturbation de la signalisation stéroïdienne et de la signalisation rétinoïdienne par l’intermédiaire d’un même récepteur (SHP ; NR0B2). Par ailleurs, ces auteurs ont également mis en évidence des effets complexes à la fois dans les cellules de Leydig et les cellules germinales. Ce travail montre la difficulté d’appréhender les effets au sein d’un tissu quand une même substance peut perturber plusieurs voies de signalisation et plusieurs types cellulaires interdépendants. Par exemple, l’implication de SHP dans la gamétogenèse n’a été proposée que très récemment (Volle et coll., 2007
).
Effets pathologiques
Les effets moléculaires et cellulaires des PE peuvent avoir pour conséquences des altérations physiologiques entraînant une pathologie au niveau des organes reproducteurs et/ou de la fonction de reproduction.
Effets des anti-androgènes
L’hypospadias et la cryptorchidie peuvent être causées par des anti-androgènes. D’autres paramètres additionnels sont utilisés pour mesurer expérimentalement les effets de substances sur l’action des androgènes au cours du développement des rongeurs mâles. Ceux-ci comprennent la distance anogénitale, le poids du muscle levator ani-bulbocaverneux, la rétention des tétines chez le mâle, le poids et la structure histologique du testicule, de l’épididyme, de la prostate ou des vésicules séminales et la production de spermatozoïdes (Gray et coll., 2001
). Le comportement lors de l’accouplement peut être utilisé pour appréhender des effets au niveau du système nerveux central.
Deux grands modes d’action ont été proposés pour les anti-androgènes. Certains ont directement une activité antagoniste au niveau du récepteur et d’autres perturbent la synthèse et/ou le métabolisme des androgènes. Ainsi, par exemple, certains phtalates peuvent inhiber la synthèse d’androgènes des cellules de Leydig fœtales de rat (Parks et coll., 2000
; Foster et coll., 2001
). Cet effet s’accompagne de malformation de l’épididyme, d’hypospadias, de la persistance de tétines (aréoles de structures mammaires) et d’une diminution de la distance anogénitale.
Effets des substances œstrogéniques
Chez le mâle, il semble exister de grande ressemblance entre l’effet des œstrogènes et celui des substances anti-androgéniques peut-être du fait que les œstrogènes peuvent diminuer le taux de récepteur aux androgènes (Williams et coll., 2001
; Sharpe et coll., 2003
). De plus, les œstrogènes peuvent inhiber l’expression de l’INSL3, qui permet la descente testiculaire.
Chez la femelle, la substance œstrogénique qui fut la mieux étudiée est le DES. Celle-ci cause des malformations des organes reproducteurs, essentiellement au niveau du vagin et de l’utérus. Les œstrogènes sont également des acteurs importants du développement de la glande mammaire et des cancers mammaires.
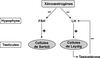
Effets systémiques
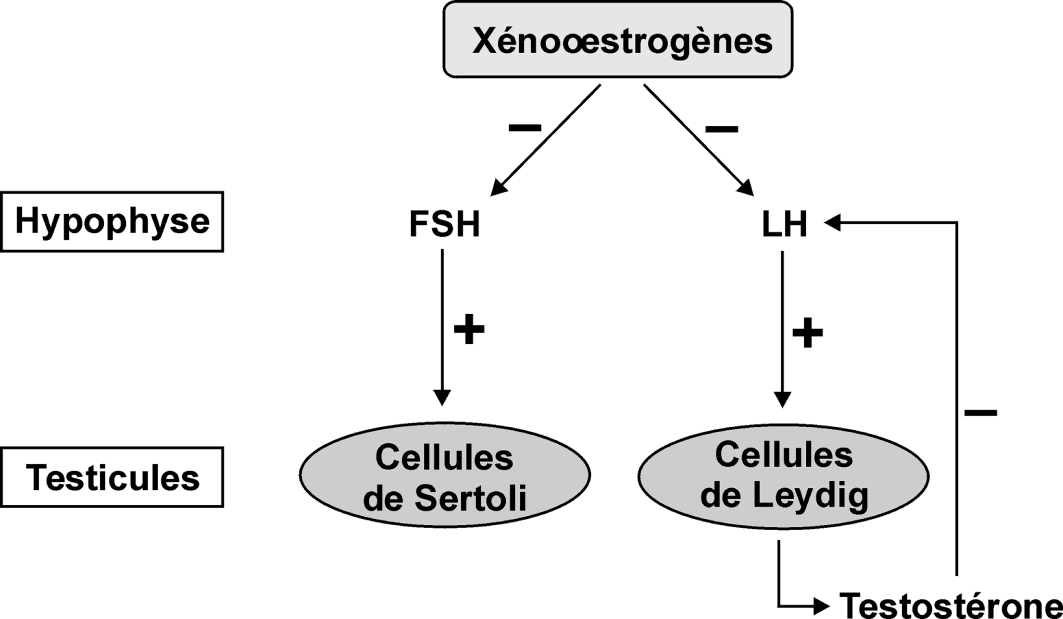
Les xéno-œstrogènes pourraient également avoir un effet au niveau systémique conduisant à la perturbation du contrôle hormonal du développement de certains organes comme le testicule. Ces substances peuvent inhiber la sécrétion des hormones hypophysaires par rétrocontrôle sur les récepteurs aux œstrogènes présents au niveau de l’hypothalamus (figure 15.3
).
L’action de ces xéno-œstrogènes durant la période critique du développement fœtal correspondant à la morphogenèse testiculaire conduit au syndrome de dysgénésie testiculaire. En effet, il est admis que l’oligospermie, le cancer du testicule, la cryptorchidie et l’hypospadias sont des manifestations d’un même syndrome de dysgénésie testiculaire (SDT). Le SDT résulte de la perturbation hormonale du programme embryonnaire de développement des gonades durant la vie fœtale. Le SDT serait ainsi la conséquence d’une exposition anormale à des facteurs environnementaux (perturbateurs endocriniens) dont les actions seraient favorisées par un terrain génétique particulier. Il faut cependant noter que le concept de dysgénésie testiculaire est contesté par certains auteurs qui pensent que les quatre manifestations cliniques (oligospermie, cancer du testicule, cryptorchidie et hypospadias) ne sont liées ni sur le plan épidémiologique ni sur celui des causes sous-jacentes (Akre et Richiardi, 2009
).
Par l’intermédiaire d’un rétrocontrôle négatif, les xéno-œstrogènes inhibent les sécrétions hypophysaires de FSH et de LH. L’induction de l’aromatase ou l’inhibition des enzymes qui dégradent l’œstradiol peut conduire à des effets cancérogènes. Les xéno-œstrogènes cumulatifs ou non sont également susceptibles de jouer un rôle prépondérant dans la régulation de la prolifération cellulaire du tissu mammaire.
Impact des polymorphismes génétiques
Divers polymorphismes dans des acteurs clefs de la fonction de reproduction sont connus. Ainsi des polymorphismes dans les gènes codant pour l’INSL3 et son récepteur RXFP2 ont été décrits chez l’être humain mais leur rôle dans la pathogenèse de la cryptorchidie reste débattu (Virtanen et Toppari, 2008
). Cependant, ces polymorphismes pourraient modifier l’activité des protéines codées par ces gènes et s’ajouter à l’effet de perturbateurs tels que le DBP qui modifie l’expression de l’INSL3. Des polymorphismes ont également été décrits pour les récepteurs aux androgènes (AR) et aux œstrogènes (ER) ainsi que pour SF1 ou pour certains PPAR. Plus globalement, la notion d’inter action entre gènes et environnement reste très peu explorée dans le cadre de la reprotoxicité. Pourtant pour certains perturbateurs endocriniens (notamment des phtalates) des effets très différents ont été rapportés d’une espèce à l’autre et parfois même entre deux lignées différentes d’une même espèce.
Effets des mélanges complexes
Parmi les études qui ont abordé les effets de mélanges complexes, il est possible de dégager deux grandes notions. La première : des « perturbateurs endocriniens de la même catégorie » ont des effets globalement additifs ou cumulatifs. Ainsi, à une dose donnée, isolés, plusieurs composés peuvent ne pas avoir d’effet mais, réunis, ceux-ci peuvent perturber le développement d’un tissu. Bien que dans une certaine mesure cet effet puisse être prévu par des modèles élaborés prenant en compte les effets des composés isolés en fonction d’une gamme complète de doses, de telles expériences soulèvent la question de l’effet des faibles doses. Une des illustrations les plus probantes qui alimente ce débat est l’effet de substances considérées comme « anti-androgéniques ». Il a été montré que la flutamide, la vinclozoline ou la prycymidone peuvent à doses faibles ne pas modifier la distance anogénitale, un critère de la masculinisation mâle mais que le mélange de ces substances diminue significativement ce paramètre (Hass et coll., 2007
). De même, chez le fœtus de rat, trois phtalates différents qui individuellement n’induisent aucune hypospadias peuvent en combinaison induire des hypospadias chez la moitié des ratons (Howdeshell et coll., 2008
). Il est donc important de connaître le mode d’action d’une substance potentiellement reprotoxique pour savoir si les effets de celle-ci risque de s’additionner à ceux d’autres perturbateurs reprotoxiques agissant par le même mode d’action.
La seconde notion qui se dégage concerne les mélanges plus complexes impliquant des perturbateurs endocriniens considérés comme de catégories différentes (avec des mécanismes d’actions différents). Dans ce cas, les effets semblent beaucoup moins prévisibles et sont moins bien compris. Un exemple de ce genre est la récente étude d’Eustache et coll. (2009
) qui ont administré un mélange de génistéine, un phyto-œstrogène, et de vinclozoline à des doses « faibles » ou « fortes » à des rats depuis la vie fœtale jusqu’à l’âge adulte. Dans ces conditions, le mélange à doses faibles peut diminuer plus fortement le nombre de spermatozoïdes que le mélange à doses fortes. Par ailleurs, à doses faibles, le gavage avec un seul de ces composés n’altère pas ce paramètre. Notons enfin, que ce type d’exposition artificielle est encore loin de reproduire la complexité de la multitude de substances potentiellement reprotoxiques auxquelles l’être humain peut être exposé. Très peu d’études ont tenté d’aborder la question de l’exposition à un mélange complexe de polluants environnementaux reflétant une situation réelle. Citons dans ce cadre le travail de Fowler et coll. (2008
) qui démontre que l’élevage de brebis gestantes sur des pâturages « fertilisés » avec des boues d’épandage altère le développement ovarien. Il semble donc qu’un mélange reflétant une exposition complexe humaine (boues provenant des égouts) puisse altérer la fertilité. Évidemment la détermination de(s) substance(s) active(s) et impliquée(s) est dans ce cas plus compliquée.
Effets à l’échelle des populations
Dans certains cas, des substances peuvent avoir des effets qui ne sont pas pathologiques à l’échelle individuelle mais sont délétères à l’échelle de la population. C’est en particulier le cas d’un effet sur le sex-ratio. À titre d’exemple, un effet possible sur le sex-ratio dans l’espèce humaine de polluants persistants, dont certains sont des perturbateurs endocriniens, tels que le plomb (Simonsen et coll., 2006
) ou la dioxine (Mocarelli et coll., 2000
) a été rapporté.
Bibliographie
[1] AKBAS GE, SONG J, TAYLOR HS. A HOXA10 estrogen response element (ERE) is differentially regulated by 17 β-estradiol and diethylstilbestrol (DES).
J Mol Biol. 2004;
340:1013
-1023

[2] AKRE O, RICHIARDI L. Does a testicular dysgenesis syndrome exist?.
Hum Reprod. 2009;
24:2053
-2060
[3] ANWAY MD, SHOW MD, ZIRKIN BR. Protein C inhibitor expression by adult rat Sertoli cells: effects of testosterone withdrawal and replacement.
J Androl. 2005;
26:578
-85
[4] ARANDA A, PASCUAL A. Nuclear hormone receptors and gene expression.
Physiol Rev. 2001;
81:1269
-1304
[5] AUBOEUF D, RIEUSSET J, FAJAS L, VALLIER P, FRERING V, et coll. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-α in humans: no alteration in adipose tissue of obese and NIDDM patients.
Diabetes. 1997;
46:1319
-1327
[6] AUHAREK SA, DE FRANCA LR, MCKINNELL C, JOBLING MS, SCOTT HM, SHARPE RM. Prenatal plus postnatal exposure to Di(n-Butyl) phthalate and/or flutamide markedly reduces final sertoli cell number in the rat.
Endocrinology. 2010;
151:2868
-2875
[7] BROMER JG, WU J, ZHOU Y, TAYLOR HS. Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming.
Endocrinology. 2009;
150:3376
-3382
[8] CASALS-CASAS C, FEIGE JN, DESVERGNE B. Interference of pollutants with PPARs: endocrine disruption meets metabolism.
Int J Obes (Lond). 2008;
32 (suppl 6):S53
-61
[9] COUSE JF, KORACH KS. Estrogen receptor-α mediates the detrimental effects of neonatal diethylstilbestrol (DES) exposure in the murine reproductive tract.
Toxicology. 2004;
205:55
-63
[10] COUSE JF, LINDZEY J, GRANDIEN K, GUSTAFSSON JA, KORACH KS. Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse.
Endocrinology. 1997;
138:4613
-4621
[11] DOUARD V, BRUNET F, BOUSSAU B, AHRENS-FATH I, VLAEMINCK-GUILLEM V, et coll. The fate of the duplicated androgen receptor in fishes: a late neofunctionalization event?.
BMC Evol Biol. 2008;
8:336
[12] ESCRIVA H, DELAUNAY F, LAUDET V. Ligand binding and nuclear receptor evolution.
Bioessays. 2000;
22:717
-727
[13] EUSTACHE F, MONDON F, CANIVENC-LAVIER MC, LESAFFRE C, FULLA Y, et coll. Chronic dietary exposure to a low-dose mixture of genistein and vinclozolin modifies the reproductive axis, testis transcriptome, and fertility.
Environ Health Perspect. 2009;
117:1272
-1279
[14] FERRARA D, HALLMARK N, SCOTT H, BROWN R, MCKINNELL C, et coll. Acute and long-term effects of in utero exposure of rats to di(n-butyl) phthalate on testicular germ cell development and proliferation.
Endocrinology. 2006;
147:5352
-5362
[15] FLAMANT F, GAUTHIER K, SAMARUT J. Thyroid hormones signaling is getting more complex: STORMs are coming.
Mol Endocrinol. 2007;
21:321
-333
[16] FORMAN BM, CHEN J, EVANS RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ.
Proc Natl Acad Sci USA. 1997;
94:4312
-4317
[17] FOSTER PM, MYLCHREEST E, GAIDO KW, SAR M. Effects of phthalate esters on the developing reproductive tract of male rats.
Hum Reprod Update. 2001;
7:231
-235
[18] FOWLER PA, DORÀ NJ, MCFERRAN H, AMEZAGA MR, MILLER DW, et coll. In utero exposure to low doses of environmental pollutants disrupts fetal ovarian development in sheep.
Mol Hum Reprod. 2008;
14:269
-80
[19] GERMAIN P, STAELS B, DACQUET C, SPEDDING M, LAUDET V. Overview of nomenclature of nuclear receptors.
Pharmacol Rev. 2006;
58:685
-704
[20] GRASSELLI F, BARATTA L, BAIONI L, BUSSOLATI S, RAMONI R, et coll. Bisphenol A disrupts granulosa cell function.
Domest Anim Endocrinol. 2010;
39:34
-39
[21] GRAY LE, OSTBY J, FURR J, WOLF CJ, LAMBRIGHT C, et coll. Effects of environmental antiandrogens on reproductive development in experimental animals.
Hum Reprod Update. 2001;
7:248
-264
[22] GRONEMEYER H, GUSTAFSSON JA, LAUDET V. Principles for modulation of the nuclear receptor superfamily.
Nat Rev Drug Discov. 2004;
3:950
-964
[23] HABERT R, MUCZYNSKI V, LEHRAIKI A, LAMBROT R, LÉCUREUIL C, et coll. Adverse effects of endocrine disruptors on the fœtal testis development: focus on the phthalates.
Folia Histochem Cytobiol. 2009;
47:S67
-S74
[24] HASS U, SCHOLZE M, CHRISTIANSEN S, DALGAARD M, VINGGAARD AM, et coll. Combined exposure to anti-androgens exacerbates disruption of sexual differentiation in the rat.
Environ Health Perspect. 2007;
115 (suppl 1):122
-128
[25] HOLSBERGER DR, COOKE PS. Understanding the role of thyroid hormone in Sertoli cell development: a mechanistic hypothesis.
Cell Tissue Res. 2005;
322:133
-140
[26] HOLSBERGER DR, KIESEWETTER SE, COOKE PS. Regulation of neonatal Sertoli cell development by thyroid hormone receptor α1.
Biol Reprod. 2005;
73:396
-403
[27] HOWDESHELL KL, RIDER CV, WILSON VS, GRAY LE JR. Mechanisms of action of phthalate esters, individually and in combination, to induce abnormal reproductive development in male laboratory rats.
Environ Res. 2008;
108:168
-176
[28] ISSEMANN I, GREEN S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators.
Nature. 1990;
347:645
-650
[29] KASTNER P, KRUST A, TURCOTTE B, STROPP U, TORA L, et coll. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B.
EMBO J. 1990;
9:1603
-1614
[30] KHALAF H, LARSSON A, BERG H, MCCRINDLE R, ARSENAULT G, OLSSON PE. Diastereomers of the brominated flame retardant 1,2-dibromo-4-(1,2 dibromoethyl)cyclohexane induce androgen receptor activation in the hepg2 hepatocellular carcinoma cell line and the lncap prostate cancer cell line.
Environ Health Perspect. 2009;
117:1853
-1859
[31] KLIEWER SA, SUNDSETH SS, JONES SA, BROWN PJ, WISELY GB, et coll. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ.
Proc Natl Acad Sci USA. 1997;
94:4318
-4323
[32] KÖRNER W, VINGGAARD AM, TÉROUANNE B, MA R, WIELOCH C, et coll. Interlaboratory comparison of four in vitro assays for assessing androgenic and antiandrogenic activity of environmental chemicals.
Environ Health Perspect. 2004;
112:695
-702
[33] KRENTZ AD, MURPHY MW, KIM S, COOK MS, CAPEL B, et coll. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency.
Proc Natl Acad Sci USA. 2009;
106:22323
-22328
[34] KUIPER GG, CARLSSON B, GRANDIEN K, ENMARK E, HÄGGBLAD J, et coll. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β.
Endocrinology. 1997;
138:863
-870
[35] KWINTKIEWICZ J, NISHI Y, YANASE T, GIUDICE LC. Peroxisome proliferator-activated receptor-gamma mediates bisphenol A inhibition of FSH-stimulated IGF-1, aromatase, and estradiol in human granulosa cells.
Environ Health Perspect. 2010;
118:400
-406
[36] LAGANIÈRE J, DEBLOIS G, GIGUÈRE V. Functional genomics identifies a mechanism for estrogen activation of the retinoic acid receptor α1 gene in breast cancer cells.
Mol Endocrinol. 2005;
19:1584
-1592
[37] LAGUË E, TREMBLAY JJ. Estradiol represses insulin-like 3 expression and promoter activity in MA-10 Leydig cells.
Toxicology. 2009;
258:101
-105
[38] LE MAIRE A, GRIMALDI M, ROECKLIN D, DAGNINO S, VIVAT-HANNAH V, et coll. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors.
EMBO Rep. 2009;
10:367
-373
[39] LE MAIRE A, BOURGUET W, BALAGUER P. A structural view of nuclear hormone receptor: endocrine disruptor interactions.
Cell Mol Life Sci. 2010;
67:1219
-1237
[40] LEHRAIKI A, RACINE C, KRUST A, HABERT R, LEVACHER C. Phthalates impair germ cell number in the mouse fetal testis by an androgen- and estrogen-independent mechanism.
Toxicol Sci. 2009;
111:372
-382
[41] LIVERA G, DELBES G, PAIRAULT C, ROUILLER-FABRE V, HABERT R. Organotypic culture, a powerful model for studying rat and mouse fetal testis development.
Cell Tissue Res. 2006;
324:507
-521
[42] MASSAAD C, BAROUKI R. An assay for the detection of xenoestrogens based on a promoter containing overlapping EREs.
Environ Health Perspect. 1999;
107:563
-536
[43] MOCARELLI P, GERTHOUX PM, FERRARI E, PATTERSON DG, JR., KIESZAK SM, et coll. Paternal concentrations of dioxin and sex ratio of offspring.
Lancet. 2000;
355:1858
-1863
[44] MORIYAMA K, TAGAMI T, AKAMIZU T, USUI T, SAIJO M, et coll. Thyroid hormone action is disrupted by bisphenol A as an antagonist.
J Clin Endocrinol Metab. 2002;
87:5185
-5190
[45] MUÑOZ-DE-TORO M, MARKEY CM, WADIA PR, LUQUE EH, RUBIN BS, et coll. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice.
Endocrinology. 2005;
146:4138
-4147
[46]NUCLEAR RECEPTORS NOMENCLATURE COMMITTEE. A unified nomenclature system for the nuclear receptor superfamily.
Cell. 1999;
97:161
-163
[47] OHTAKE F, BABA A, TAKADA I, OKADA M, IWASAKI K, et coll. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase.
Nature. 2007;
446:562
-566
[48] PARIS F, BALAGUER P, TÉROUANNE B, SERVANT N, LACOSTE C, et coll. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit α and β estrogen activities and antiandrogen activity in reporter cell lines.
Mol Cell Endocrinol. 2002;
193:43
-49
[49] PARKS LG, OSTBY JS, LAMBRIGHT CR, ABBOTT BD, KLINEFELTER GR, et coll. The plasticizer diethylhexyl phthalate induces malformations by decreasing fetal testosterone synthesis during sexual differentiation in the male rat.
Toxicol Sci. 2000;
58:339
-349
[50] PETTERSSON K, DELAUNAY F, GUSTAFSSON JA. Estrogen receptor β acts as a dominant regulator of estrogen signaling.
Oncogene. 2000;
19:4970
-1978
[51] SASSI-MESSAI S, GIBERT Y, BERNARD L, NISHIO S, FERRI LAGNEAU KF, et coll. The phytoestrogen genistein affects zebrafish development through two different pathways.
PLoS One. 2009;
4:e4935
[52] SHANG Y, BROWN M. Molecular determinants for the tissue specificity of SERMs.
Science. 2002;
295:2465
-2468
[53] SHARPE RM, RIVAS A, WALKER M, MCKINNELL C, FISHER JS. Effect of neonatal treatment of rats with potent or weak (environmental) œstrogens, or with a GnRH antagonist, on Leydig cell development and function through puberty into adulthood.
Int J Androl. 2003;
26:26
-36
[54] SHI Y, HON M, EVANS , . The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling.
Proc Natl Acad Sci USA. 2002;
99:2613
-2618
[55] SIMONSEN CR, ROGE R, CHRISTIANSEN U, LARSEN T, BONDE JP. Effects of paternal blood lead levels on offspring sex ratio.
Reprod Toxicol. 2006;
295:3
-4
[56] SUN H, SHEN OX, WANG XR, ZHOU L, ZHEN SQ, CHEN XD. Anti-thyroid hormone activity of bisphenol A, tetrabromobisphenol A and tetrachlorobisphenol A in an improved reporter gene assay.
Toxicol In Vitro. 2009;
23:950
-954
[57] SWEDENBORG E, POWER KA, CAI W, PONGRATZ I, RÜEGG J. Regulation of estrogen receptor β activity and implications in health and disease.
Cell Mol Life Sci. 2009;
66:3873
-3894
[58] VIRTANEN HE, TOPPARI J. Epidemiology and pathogenesis of cryptorchidism.
Hum Reprod Update. 2008;
14:49
-58
[59] VOLLE DH, MOUZAT K, DUGGAVATHI R, SIDDEEK B, DÉCHELOTTE P, et coll. Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility.
Mol Endocrinol. 2007;
21:1014
-1027
[60] VOLLE DH, DECOURTEIX M, GARO E, MCNEILLY J, FENICHEL P, et coll. The orphan nuclear receptor small heterodimer partner mediates male infertility induced by diethylstilbestrol in mice.
J Clin Invest. 2009;
119:3752
-3764
[61] WILLIAMS K, FISHER JS, TURNER KJ, MCKINNELL C, SAUNDERS PT, SHARPE RM. Relationship between expression of sex steroid receptors and structure of the seminal vesicles after neonatal treatment of rats with potent or weak estrogens.
Environ Health Perspect. 2001;
109:1227
-1235
→ Aller vers SYNTHESE