Bisphénol A
2011
| ANALYSE |
30-
Relation structure–fonction
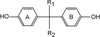
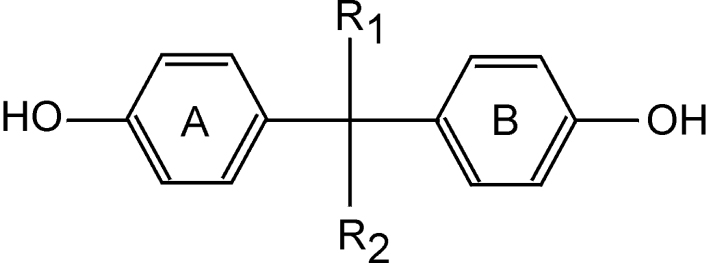
La structure chimique d’une classe de perturbateurs endocriniens, les diphénylalcanes hydroxylés ou bisphénols (BP), ayant une activité œstrogénique (œstrogéno-mimétique) significative, est composée par deux cycles aromatiques (phényles) liés par un pont carbone. La structure du bisphénol A (BPA, 4,4’-dihydroxy-2,2-diphénylpropane en nomenclature IUPAC), composé le plus connu de cette famille, est représentée en figure 30.1 .
.
.
Ces molécules interagissent avec les mêmes récepteurs (ERα, ERβ ou ERRγ) que les œstrogènes naturels comme l’œstradiol (E2) (Agatonovic-Kustrin et Turner, 2008). Par différence avec l’œstradiol, la structure d’un récepteur cristallisé avec le BP n’a pas été résolue par analyse aux rayons X. Cependant, des études expérimentales et/ou théoriques ont mis en évidence différents types d’interactions substrat (BP)/récepteur, mettant en œuvre soit des analogies avec E2 (groupements hydrophobes, hydrophiles, accepteurs/donneurs de liaison H), soit des simulations atomistiques. Ces études ont conduit à des relations bien définies entre les caractéristiques structurales et l’activité biologique (souvent in vitro) de cette classe de BP.
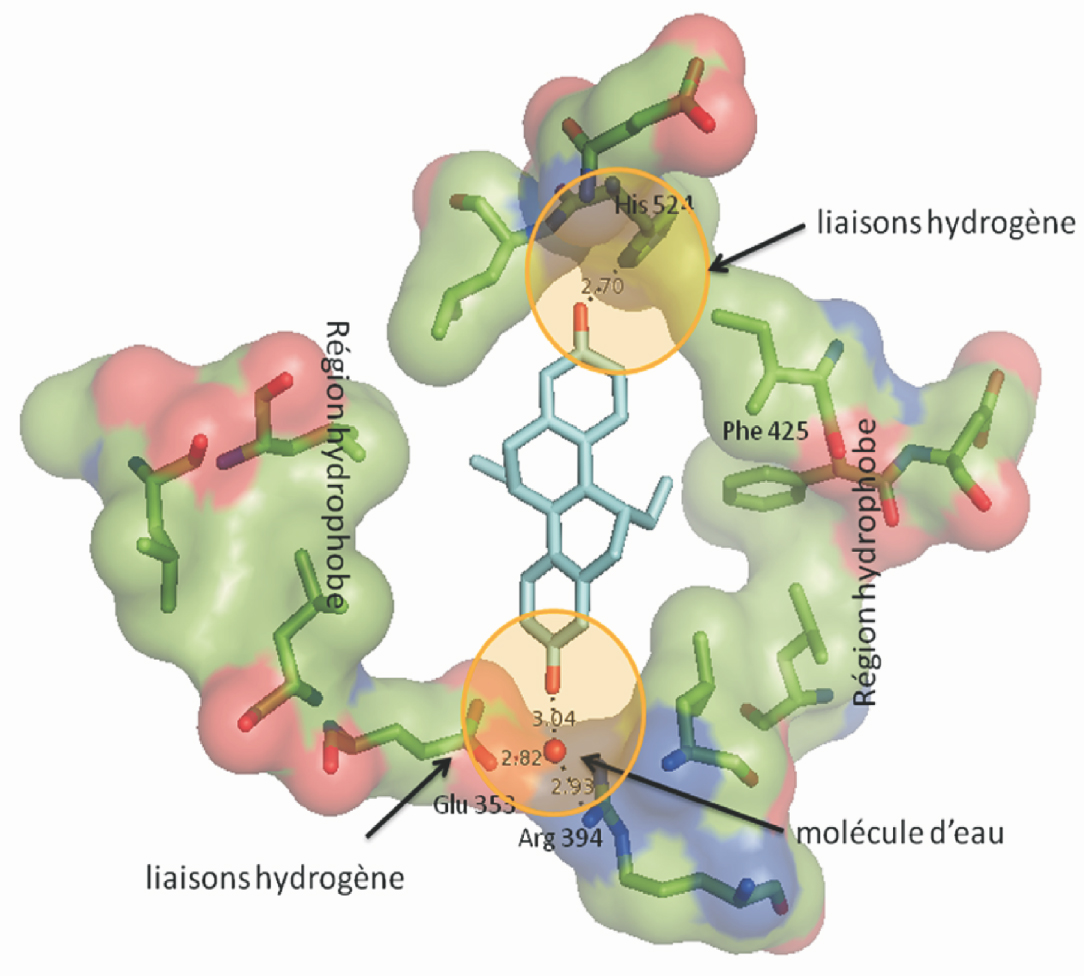
). Par différence avec l’œstradiol, la structure d’un récepteur cristallisé avec le BP n’a pas été résolue par analyse aux rayons X. Cependant, des études expérimentales et/ou théoriques ont mis en évidence différents types d’interactions substrat (BP)/récepteur, mettant en œuvre soit des analogies avec E2 (groupements hydrophobes, hydrophiles, accepteurs/donneurs de liaison H), soit des simulations atomistiques. Ces études ont conduit à des relations bien définies entre les caractéristiques structurales et l’activité biologique (souvent in vitro) de cette classe de BP.En effet, la partie 66-Kd du récepteur ER se compose de trois domaines structuralement distincts : un domaine N-terminal de liaison à l’ADN, un domaine N-terminal et un domaine C-terminal formant une poche hydrophobe (Brzozowski et coll., 1997 ; Tanenbaum et coll., 1998). Cette cage tridimensionnelle est composée de plusieurs sites de liaison non spécifiques sur lesquels peuvent se loger l’œstrogène ainsi que d’autres ligands (figure 30.2). Avec un volume de 440 Å3, la poche de liaison est beaucoup plus grande qu’une molécule d’œstrogène, occupant à elle seule un volume de 245 Å3 (pour E2). Ce volume vide est construit à l’aide de résidus non polaires lui conférant la capacité à se lier avec une grande variété de ligands dont la structure prend en compte un groupement phénol, discriminant retrouvé sur la molécule d’œstrogène.
; Tanenbaum et coll., 1998). Cette cage tridimensionnelle est composée de plusieurs sites de liaison non spécifiques sur lesquels peuvent se loger l’œstrogène ainsi que d’autres ligands (figure 30.2). Avec un volume de 440 Å3, la poche de liaison est beaucoup plus grande qu’une molécule d’œstrogène, occupant à elle seule un volume de 245 Å3 (pour E2). Ce volume vide est construit à l’aide de résidus non polaires lui conférant la capacité à se lier avec une grande variété de ligands dont la structure prend en compte un groupement phénol, discriminant retrouvé sur la molécule d’œstrogène. | Figure 30.2 Structure à rayons X de la poche hydrophobe du ERα avec le substrat naturel 17β œstradiol (E2) (Brzozowski et coll., 1997 ; Tanenbaum et coll., 1998) |
Études in-silico
Au cours de ces dernières années, un grand nombre de travaux portant sur des méthodes QSAR ont été dédiés à l’étude des interactions de ligands non stéroïdiens avec le récepteur ER. Le BPA et ses dérivées ont fait l’objet de quelques travaux spécifiques (Coleman et coll., 2003) mais sont le plus souvent inclus dans le jeu d’entraînement ou de validation des modèles QSAR (Gao et coll., 1999 ; Shi et coll., 2001 ; Waller, 2004 ; Devillers et coll., 2006 ; Kadowaki et coll., 2007 ; Liu et coll., 2007 ; Roncaglioni et coll., 2008a et b). Il faut toutefois souligner que dans la plupart de ces travaux, le critère d’évaluation est l’affinité de liaison au récepteur, absolue (binding affinity, BA) ou relative au ligand naturel E2 (relative binding affinity, RBA). Des critères d’évaluation plus complexes sont rarement considérés (Coleman et coll., 2003 ; Roncaglioni et coll., 2008a).
) mais sont le plus souvent inclus dans le jeu d’entraînement ou de validation des modèles QSAR (Gao et coll., 1999 ; Shi et coll., 2001 ; Waller, 2004 ; Devillers et coll., 2006 ; Kadowaki et coll., 2007 ; Liu et coll., 2007 ; Roncaglioni et coll., 2008a et b). Il faut toutefois souligner que dans la plupart de ces travaux, le critère d’évaluation est l’affinité de liaison au récepteur, absolue (binding affinity, BA) ou relative au ligand naturel E2 (relative binding affinity, RBA). Des critères d’évaluation plus complexes sont rarement considérés (Coleman et coll., 2003 ; Roncaglioni et coll., 2008a).Les jeux d’entraînements sont généralement composés de plusieurs centaines de molécules, mais les jeux de validation sont souvent plus petits (une centaine de molécules), sauf dans quelques cas particuliers (Liu et coll., 2007). Ces modèles permettent la détermination qualitative et/ou quantitative de l’activité œstrogénique en termes d’affinité. Des accords allant jusqu’à 90 % ont été obtenus sur des modèles qualitatifs fournissant une réponse binaire, de type active ou non active (Liu et coll., 2007). Les modèles quantitatifs, plus complexes, présentent également de très bonnes corrélations (R2>0,90) en ce qui concerne l’affinité avec les ER, et des pouvoirs prédictifs (q2>0,6) pour de très grands ensembles de ligands de structures extrêmement variées (Shi et coll., 2001) (figure 30.3).
). Ces modèles permettent la détermination qualitative et/ou quantitative de l’activité œstrogénique en termes d’affinité. Des accords allant jusqu’à 90 % ont été obtenus sur des modèles qualitatifs fournissant une réponse binaire, de type active ou non active (Liu et coll., 2007). Les modèles quantitatifs, plus complexes, présentent également de très bonnes corrélations (R2>0,90) en ce qui concerne l’affinité avec les ER, et des pouvoirs prédictifs (q2>0,6) pour de très grands ensembles de ligands de structures extrêmement variées (Shi et coll., 2001) (figure 30.3).Une amélioration significative a été apportée en couplant les modèles QSPR traditionnels avec des approches de type « docking » et ab-initio.
Dans ce cas, le modèle se caractérise par une excellente corrélation (R2=0,991) et une très bonne prédictivité (q2=0,92) (Sippl et coll., 2000).
).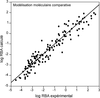 | Figure 30.3 Exemple d’une corrélation linéaire (R2=0,91) entre l’affinité de liaison relative à E2 (relative binding affinity) calculée et expérimentale (Shi et coll., 2001) |
Malheureusement, seul un ensemble restreint de molécules (30) a été considéré, et ceci principalement à cause des limitations informatiques. Par ailleurs, des travaux de type QSAR traditionnel ont été effectués dans le cadre des normes réglementaires OCDE (Tong et coll., 2003 ; Liu et coll., 2006 ; Jensen et coll., 2008). Ces modèles ont également permis d’identifier les principales caractéristiques structurales nécessaires lors de l’interaction ligand récepteur. Néanmoins, des informations plus détaillées sur les interactions microscopiques entre le ligand et l’ER peuvent être déterminées en combinant des approches expérimentales dans lesquelles plusieurs ligands sélectivement modifiés sont testés à l’aide de modèles QSAR, de docking ou d’atomistique. En particulier, l’étude des interactions ligand-ER portant sur des modèles de chimie quantique a montré que l’énergie d’interaction théorique et l’affinité déterminée de façon expérimentale sont bien corrélées (Fukuzawa et coll., 2005). Ces modèles donnent non seulement des informations sur les mécanismes sous-jacents, mais également une relation linéaire (R>0,8) entre l’énergie d’interaction calculée et l’activité expérimentale. Le croisement des données théoriques avec les informations expérimentales a permis l’individualisation des caractéristiques structurales du ligand (Kadowaki et coll., 2007).
; Liu et coll., 2006 ; Jensen et coll., 2008). Ces modèles ont également permis d’identifier les principales caractéristiques structurales nécessaires lors de l’interaction ligand récepteur. Néanmoins, des informations plus détaillées sur les interactions microscopiques entre le ligand et l’ER peuvent être déterminées en combinant des approches expérimentales dans lesquelles plusieurs ligands sélectivement modifiés sont testés à l’aide de modèles QSAR, de docking ou d’atomistique. En particulier, l’étude des interactions ligand-ER portant sur des modèles de chimie quantique a montré que l’énergie d’interaction théorique et l’affinité déterminée de façon expérimentale sont bien corrélées (Fukuzawa et coll., 2005). Ces modèles donnent non seulement des informations sur les mécanismes sous-jacents, mais également une relation linéaire (R>0,8) entre l’énergie d’interaction calculée et l’activité expérimentale. Le croisement des données théoriques avec les informations expérimentales a permis l’individualisation des caractéristiques structurales du ligand (Kadowaki et coll., 2007).Relation structure-fonction
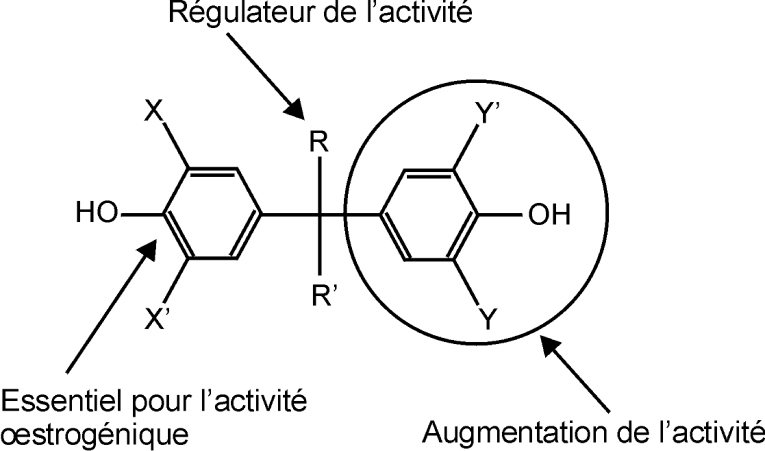
Les ERs peuvent lier un nombre important de molécules structuralement différentes. Les caractéristiques principales de leurs ligands naturels (stéroïdes) relèvent de la présence d’un groupement aromatique hydroxylé (cycle phénolique) et d’un squelette hydrophobe (Anstead et coll., 1997 ; Fang et coll., 2001). Ces caractéristiques essentielles pour l’activité œstrogénique se retrouvent également dans les BP, molécules caractérisées par un cycle aromatique porteur d’un groupement hydroxyle (cycle A, figure 30.1) et éventuellement substituée en position para. La structure à cycles condensés, retrouvée dans le cas de E2 n’est par conséquent pas nécessaire pour obtenir une activité œstrogénique en terme de prolifération des cellules de cancer du sein humaines MCF-7 (Dodds et Lawson, 1936 ; Perez et coll., 1998), et peut même donner lieu à des composés non actifs comme les dérivés naphtols (Soto et coll., 1995). La présence du groupement hydroxyle est fondamentale dans l’activité œstrogénique. En effet, l’élimination d’un groupement OH conduit au 4-a-cumylphénol (HOC6H4-C(CH3)2-C6H5), composé ayant une affinité de liaison (binding affinity) avec le récepteur égale à celle du BPA, alors que l’élimination du second OH, donne lieu au composé 2,2-diphénylpropane (C6H5-C(CH3)2-C6H5), molécule complètement inactive (vis-à-vis du récepteur ERRγ) (Okada et coll., 2008).
; Fang et coll., 2001). Ces caractéristiques essentielles pour l’activité œstrogénique se retrouvent également dans les BP, molécules caractérisées par un cycle aromatique porteur d’un groupement hydroxyle (cycle A, figure 30.1) et éventuellement substituée en position para. La structure à cycles condensés, retrouvée dans le cas de E2 n’est par conséquent pas nécessaire pour obtenir une activité œstrogénique en terme de prolifération des cellules de cancer du sein humaines MCF-7 (Dodds et Lawson, 1936 ; Perez et coll., 1998), et peut même donner lieu à des composés non actifs comme les dérivés naphtols (Soto et coll., 1995). La présence du groupement hydroxyle est fondamentale dans l’activité œstrogénique. En effet, l’élimination d’un groupement OH conduit au 4-a-cumylphénol (HOC6H4-C(CH3)2-C6H5), composé ayant une affinité de liaison (binding affinity) avec le récepteur égale à celle du BPA, alors que l’élimination du second OH, donne lieu au composé 2,2-diphénylpropane (C6H5-C(CH3)2-C6H5), molécule complètement inactive (vis-à-vis du récepteur ERRγ) (Okada et coll., 2008).La présence d’un second groupement fonctionnel à base d’oxygène (hydroxyle, cétone, ou carbonyle), positionné sur la partie terminale de la molécule, en opposition au cycle phénolique, confère ou du moins augmente l’activité œstrogénique (cellules MCF-7) (Perez et coll., 1998). Le BPA et le bisphénol F (BPF, 4,4’-Methylenbisphénol), tous deux porteurs de deux groupements OH, ou le bisphénol A dimethacrylate (BIS-DMA) et le bisphénol-A-bis chloroformate (BPACF) ont montré une activité œstrogénique lors des tests de prolifération et d’induction protéique (Perez et coll., 1998).
Par analogie avec E2, ces deux groupements sont nécessaires pour établir deux interactions liantes de type liaison H à l’intérieur de la poche de E2 (voir figure 30.2) avec d’un côté, une molécule d’eau, de Glu353 et de Arg394, et de l’autre, une molécule de His 524 (Brzozowski et coll., 1997 ; Tanenbaum et coll., 1998). Néanmoins, la présence et la distance entre ces deux groupements hydroxyles ne sont pas les seuls facteurs critiques. En particulier, la nature des substituants liés au pont carbone peut déterminer l’activité œstrogénique. Par exemple, la substitution des groupements méthyles positionnés sur ce dernier, par des groupements hydrophiles provoque une baisse de l’activité œstrogénique, causée par la diminution de la force d’interaction avec la partie hydrophobe du récepteur (Kitamura et coll., 2005). Une autre expérience a montré que le remplacement des groupements CH3 par des groupements CF3, donnant lieu au bisphénol AF (BAF, 2,2-Bis(4-hydroxyphenyl) hexafluoropropane), induit une baisse de l’activité (Okada et coll., 2008). En revanche, le remplacement des deux groupements méthyles par des chaînes propylées accroît l’expression des gènes et des protéines (Matsushima et coll., 2010).
) avec d’un côté, une molécule d’eau, de Glu353 et de Arg394, et de l’autre, une molécule de His 524 (Brzozowski et coll., 1997 ; Tanenbaum et coll., 1998). Néanmoins, la présence et la distance entre ces deux groupements hydroxyles ne sont pas les seuls facteurs critiques. En particulier, la nature des substituants liés au pont carbone peut déterminer l’activité œstrogénique. Par exemple, la substitution des groupements méthyles positionnés sur ce dernier, par des groupements hydrophiles provoque une baisse de l’activité œstrogénique, causée par la diminution de la force d’interaction avec la partie hydrophobe du récepteur (Kitamura et coll., 2005). Une autre expérience a montré que le remplacement des groupements CH3 par des groupements CF3, donnant lieu au bisphénol AF (BAF, 2,2-Bis(4-hydroxyphenyl) hexafluoropropane), induit une baisse de l’activité (Okada et coll., 2008). En revanche, le remplacement des deux groupements méthyles par des chaînes propylées accroît l’expression des gènes et des protéines (Matsushima et coll., 2010). L’importance du cycle benzénique B peut être évaluée en le remplaçant par des groupements alkyles. En effet, sa substitution par des groupements méthyles ou éthyles dérivés (4-tert-butylphénol et 4-tert-amylphénol) se matérialise par une baisse significative de l’activité, devenant ainsi inférieure à celle du 4-α-cumylphénol. Pour des chaines non volumineuses, l’interaction reste toutefois marquée (Okada et coll., 2008). Par conséquent, la présence d’un cycle aromatique suggère une interaction de type π-π ou XH-π (X=N,O,C) avec la poche du récepteur, interaction absente dans le cas de substituants alkyls. Enfin, la présence de substituants chlorés en position 3 ou 5 sur le cycle A a également un effet significatif sur l’activité, et permet de renforcer l’affinité de liaison avec le récepteur (Liu et coll., 2007). En effet, des dérivés chlorés peuvent se former lors du recyclage du papier thermique (par blanchissage à l’hypochlorite de sodium) qui sont 28 fois plus œstrogéniques que le BPA (Fukazawa et coll., 2002). Les requis structuraux sont résumés dans la figure 30.4.
). Par conséquent, la présence d’un cycle aromatique suggère une interaction de type π-π ou XH-π (X=N,O,C) avec la poche du récepteur, interaction absente dans le cas de substituants alkyls. Enfin, la présence de substituants chlorés en position 3 ou 5 sur le cycle A a également un effet significatif sur l’activité, et permet de renforcer l’affinité de liaison avec le récepteur (Liu et coll., 2007). En effet, des dérivés chlorés peuvent se former lors du recyclage du papier thermique (par blanchissage à l’hypochlorite de sodium) qui sont 28 fois plus œstrogéniques que le BPA (Fukazawa et coll., 2002). Les requis structuraux sont résumés dans la figure 30.4.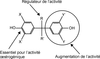
En conclusion, les bisphénols interagissent avec les mêmes récepteurs (ERα, ERβ ou ERRγ) que les œstrogènes naturels comme l’œstradiol (E2). Des études ont montré qu’il existe des relations bien définies entre les caractéristiques structurales et l’activité biologique des bisphénols. Des modèles QSAR permettent la détermination qualitative et/ou quantitative de l’activité œstrogénique en termes d’affinité et donnent des informations sur les mécanismes sous-jacents.
La présence d’un groupement aromatique hydroxylé (cycle phénolique) et d’un squelette hydrophobe, caractéristiques essentielles pour l’activité œstrogénique, se retrouvent dans les bisphénols. La présence du groupement hydroxyle est fondamentale pour l’activité œstrogénique. La présence d’un second groupement fonctionnel à base d’oxygène (hydroxyle, cétone, ou carbonyle), positionné sur la partie terminale de la molécule, en opposition au cycle phénolique, confère ou du moins augmente l’activité œstrogénique. C’est le cas du BPA et du bisphénol F (BPF, 4,4’-Methylenbisphénol) du bisphénol A dimethacrylate (BIS-DMA) et du bisphénol-A-bis chloroformate (BPACF). Ces substances montrent une activité œstrogénique lors des tests de prolifération et d’induction protéique.
Par ailleurs, la nature des substituants liés au pont carbone peut déterminer l’activité œstrogénique. Par exemple, la substitution des groupements méthyles positionnés sur ce dernier, par des groupements hydrophiles provoque une baisse de l’activité œstrogénique, causée par la diminution de la force d’interaction avec la partie hydrophobe du récepteur. La substitution du cycle benzénique B par des groupements méthyles ou éthyles dérivés (4-tert-butylphénol et 4-tert-amylphénol) se traduit par une baisse significative de l’activité. En revanche, le remplacement des groupements CH3 par des groupements CF3 (bisphénol AF) ou encore le remplacement des deux groupements méthyles par des chaînes propylées accroît l’expression des gènes et des protéines.
La présence de substituants chlorés en position 3 ou 5 sur le cycle A a également un effet significatif sur l’activité. Les dérivés chlorés qui peuvent se former lors du recyclage du papier thermique (par blanchissage à l’hypochlorite de sodium) sont 28 fois plus œstrogéniques que le BPA.
Bibliographie
[1] AGATONOVIC-KUSTRIN S, TURNER JV. Molecular structural characteristics of estrogen receptor modulators as determinants of estrogen receptor selectivity.
Mini-Rev Med Chem. 2008;
8:943- 951
[2] ANSTEAD GM, CARLSON KE, KATZENELLENBOGEN JA. The estradiol pharmacophore : ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site.
Steroids. 1997;
62:268- 303
[3] BRZOZOWSKI A, PIKE A, DAUTER Z, HUBBARD R, BONN T, et coll. Molecular basis of agonism and antagonism in the œstrogen receptor.
Nature. 1997;
389:753- 758
[4] COLEMAN KP, TOSCANO WA, WIESE TE. QSAR Models of the in vitro Estrogen Activity of Bisphenol A Analogs.
QSAR Comb Sci. 2003;
22:78- 88
[5] DEVILLERS J, MARCHAND-GENESTE N, CARPY A, PORCHER JM. SAR and QSAR modeling of endocrine disruptors.
SAR QSAR Env Res. 2006;
17:393- 412
[6] DODDS EC, LAWSON W. Synthetic estrogens without the phenanthrene nucleus.
Nature. 1936;
137:996
[7] FANG H, TONG W, SHI LM, BLAIR R, PERKINS R, et coll. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens.
Chem Res Toxicol. 2001;
14:280- 294
[8] FUKAZAWA H, WATANABE M, SHIRAISHI H, SHIOZAWA T, MATSUSHITA H, et coll. Formation of Chlorinated Derivatives of Bisphenol A in Waste Paper Recycling Plants and Their Estrogenic Activities.
J Health Sci. 2002;
48:242- 249
[9] FUKUZAWA K, KITAURA K, UEBAYASI M, NAKATA K, KAMINUMA T, NAKANO T. Ab initio quantum mechanical study of the binding energies of human estrogen receptor alpha with its ligands : an application of fragment molecular orbital method.
Comput Chem. 2005;
26:1- 10
[10] GAO H, KATZENELLENBOGEN JA, GARG R, HANSCH C. Comparative QSAR analysis of estrogen receptor ligands.
Chem Rev. 1999;
99:723
[11] JENSEN GE, NIEMELÄ JR, WEDEBYE EB, NIKOLOV NG. QSAR models for reproductive toxicity and endocrine disruption in regulatory use--a preliminary investigation.
SAR QSAR Env Res. 2008;
9:631- 641
[12] KADOWAKI T, WHEELOCK CE, ADACHI T, KUDO T, OKAMOTO S, et coll. Identification of endocrine disruptor biodegradation by integration of structure-activity relationship with pathway analysis.
Environ Sci Technol. 2007;
41:7997- 8003
[13] KITAMURA S, SUZUKI T, SANOH S, KOHTA R, JINNO N, et coll. Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds.
Tox Sc. 2005;
84:249- 259
[14] LIU H, PAPA E, GRAMATICA P. QSAR prediction of estrogen activity for a large set of diverse chemicals under the guidance of OECD principles.
Chem Res Toxicol. 2006;
19:1540- 1548
[15] LIU H, PAPA E, WALKER J, GRAMATICA P. In silico screening of estrogen-like chemicals based on different nonlinear classification models.
Mol Grap Mod. 2007;
26:135- 144
[16] OKADA H, TOKUNAGA T, LIU X, TAKAYANAGI S, MATSUSHIMA A, SHIMOHIGASHI Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-gamma.
Env Health Persp. 2008;
116:32- 38
[17] RONCAGLIONI A, BENFENATI E. In silico-aided prediction of biological properties of chemicals : œstrogen receptor-mediated effects.
Chem Soc Rev. 2008;
37:441- 450
[18] RONCAGLIONI A, PICLIN N, PINTORE M, BENFENATI E. Binary classification models for endocrine disrupter effects mediated through the estrogen receptor.
SAR QSAR Env Res. 2008;
19:697- 733
[19] SHI L, FANG H, TONG W, WU J, PERKINS R, et coll. QSAR models using a large diverse set of estrogens.
J Chem Inf Comput Sci. 2001;
41:186
[20] SIPPL W. Receptor-based 3D QSAR analysis of estrogen receptor ligands--merging the accuracy of receptor-based alignments with the computational efficiency of ligand-based methods.
Comp Aid Mo Des. 2000;
14:559- 572
[21] SOTO A, SONNENSCHEIN C, CHUNG K, FERNANDEZ MF, OLEA N, SERRANO FO. The E-SCREEN assay as a tool to identify estrogens : an update on estrogenic environmental pollutants.
Environ Health Perspect. 1995;
103:113- 122
[22] TANENBAUM DM, WANG Y, WILLIAMS SP, SIGLER PB. Crystallographic comparison of the estrogen and progesterone receptorʼs ligand binding domains.
Proc Natl Acad Sci USA. 1998;
95:5998- 6003
[23] TONG W, FANG H, HONG H, XIE Q, PERKINS R et coll. Regulatory application of SAR/QSAR for priority setting of endocrine disruptors : A perspective.
Pure Appl Chem. 2003;
75:2375- 2388
[24] WALLER CLJ. A comparative QSAR study using CoMFA, HQSAR, and FRED/SKEYS paradigms for estrogen receptor binding affinities of structurally diverse compounds.
Chem Inf Comput Sci. 2004;
44:758- 765
→ Aller vers SYNTHESE