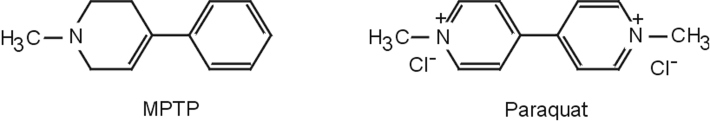III. Toxicologie
2013
23-
Mécanismes d’action neurotoxique des pesticides
De nombreuses études ont montré que plusieurs pesticides exercent une action neurotoxique chez l’Homme. Ansi une neurotoxicité (manifestations neurocomportementales, mnésiques…) a été observée lors d’intoxication aiguë (par exemple : par des organophosphorés, des organochlorés, des carbamates ou des pyréthrinoïdes). L’effet de basses concentrations de pesticides chez des populations exposées chroniquement est plus difficile à appréhender, notamment en raison des difficultés à modéliser ce type d’exposition. À titre d’exemple, les cultures cellulaires ou l’utilisation d’animaux de laboratoire ne permettent que difficilement de rechercher des effets à long terme. Des études épidémiologiques ont néanmoins permis de retrouver fréquemment un lien potentiel entre l’exposition à certains pesticides et la maladie de Parkinson, bien que de nombreuses données concernant par exemple la durée ou l’intensité de l’exposition restent imprécises.
Dans ce chapitre, une première partie est consacrée à la maladie de Parkinson et aux mécanismes cellulaires et moléculaires associés à la neurodégénérescence. Les mécanismes conduisant à la mort d’une sous-population de neurones, mécanisme essentiel dans la progression de la maladie de Parkinson, sont présentés de façon à offrir une vision générale des voies de signalisation les plus importantes. Le rôle des pesticides est ensuite détaillé par la présentation des données expérimentales les plus récentes. La deuxième partie est consacrée à l’influence des pesticides dans d’autres pathologies neuronales (dégénératives ou comportementales).
Maladie de Parkinson : mécanismes cellulaires et moléculaires associés
Spécificité des cellules dopaminergiques et rôle du stress oxydant
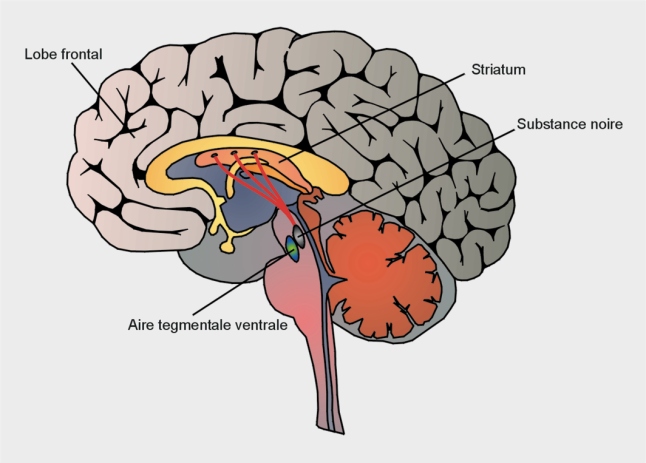
La maladie de Parkinson résulte de la dégénérescence progressive de neurones dopaminergiques de la
pars compacta localisée dans la substance noire (figure 23.1

).
http://www‑etud.iro.umontreal.ca/~rivestfr/wordpress/2008/09/19/ganglions‑de‑la‑base‑et‑systeme‑dopaminergique/ [lien obsolète]
Ceux-ci projettent leurs axones dans le striatum qui joue un rôle essentiel dans la coordination motrice. La dégénérescence de ces neurones explique plusieurs symptômes caractérisant la maladie comme l’instabilité posturale. La maladie de Parkinson se déclare quand plus de 50 % des neurones dopaminergiques de la substance noire et 75 % du contenu en dopamine au niveau striatal, ont disparu. Cette sensibilité particulière aux processus de dégénérescence s’expliquerait par les propriétés intrinsèques de ces neurones
2
Le cerveau est, à la base, particulièrement sensible au stress oxydant en raison de sa consommation importante de dioxygène (O2) (25 % de la consommation du corps pour un poids de 2 %), de son contenu important en fer (catalyseur de stress oxydant) et en lipides insaturés (cibles de peroxydation) et d’un faible niveau d’expression des enzymes de détoxication (le niveau d’expression de la catalase est seulement de 10‑20 %par rapport à celui du cœur ou du foie). La substance noire est une des zones les plus sensibles sur la base de ces caractéristiques.
qui seraient dans un état basal de stress oxydant (Richardson et coll., 2005
) lié à :
• une production intrinsèque de dérivés réactifs de l’oxygène (DRO) au cours du métabolisme de la dopamine, soit par auto-oxydation, soit par la réaction catalysée par la monoamine oxydase B (MAO-B) (Foley et Riederer, 2000
) ;
• une concentration plus faible d’antioxydants (glutathion) et une expression basse de plusieurs enzymes antioxydantes avec pour conséquence directe, un seuil de sensibilité plus bas en cas d’augmentation de la concentration en DRO (Bharath et coll., 2002
).
Dans les cerveaux post-mortem de personnes décédées de maladie de Parkinson, une augmentation des marqueurs du stress oxydant est observée (adduits sur l’ADN, peroxydation des protéines et des lipides). Ces oxydations peuvent être détectées expérimentalement avant la formation des corps de Lewy (agrégats cytoplasmiques). Or, de nombreux pesticides entraînent un stress oxydant dans plusieurs modèles testés (Banerjee et coll., 2001
).
Mécanismes associés à la neurodégénérescence
Dysfonctionnement mitochondrial
Cet aspect sera traité dans la partie consacrée aux pesticides.
Formation d’agrégats cytoplasmiques
Sur le plan histologique, les neurones dopaminergiques de patients atteints de maladie de Parkinson présentent des inclusions cytoplasmiques caractéristiques appelées « corps de Lewy » contenant des agrégats protéiques de parkine, d’ubiquitine ou d’alpha-synucléine qui est le composant principal. Les corps de Lewy apparaissent dans d’autres zones du cerveau (par exemple : cortex) avec la progression de la maladie.
Excitotoxicité glutamatergique
Le glutamate est le principal neurotransmetteur excitateur du cerveau, qui, en cas d’excès, peut causer une toxicité sur ses neurones cibles. Une déplétion en ATP (adénosine triphosphate) due à un dysfonctionnement mitochondrial (inhibition d’un ou plusieurs complexes de la chaîne respiratoire) peut conduire à cette excitotoxicité. En effet, cette diminution en ATP limite l’activité de la pompe membranaire Na
+/K
+ ATPase, essentielle au maintien de la polarité de la membrane plasmique. Une dépolarisation membranaire entraîne la diminution du blocage par le glutamate des récepteurs NMDA
3
Les récepteurs non‑NMDA pourraient aussi intervenir (Heath et Shaw, 2002
).
dépendants du voltage qui sont aussi des canaux calciques. Cette levée d’inhibition facilite l’ouverture et le passage des ions calciques. Un influx trop élevé de Ca
2+ peut ainsi conduire à la mort des cellules dopaminergiques (Greenamyre et coll., 1999
) (figure 23.2
).
Mort cellulaire
La nature de la mort cellulaire de ces neurones fait l’objet d’un débat. Il est possible que l’apoptose, la nécrose et l’autophagie puissent toutes intervenir (Foley et Riederer, 2000
). L’analyse de cerveaux post-mortem de patients atteints de maladie de Parkinson montre une activation de la caspase 3 suggérant une implication de l’apoptose mais des signes d’autophagie sont également détectés (Hartmann et coll., 2000
; Kroemer et Blomgren, 2007
) (figure 23.2
).
Maladie de Parkinson : composantes génétique et environnementale
Cette maladie est majoritairement sporadique. Quelques formes sont monogéniques, c’est-à-dire liées à la mutation d’un gène unique. La caractérisation des gènes impliqués (à forte pénétrance) et de leurs fonctions permet de mieux comprendre les mécanismes cellulaires pathogéniques
4
Bien que l’importance d’un dysfonctionnement de ces gènes dû à une variation d’expression et/ou d’activité, fasse l’objet de controverses pour les formes sporadiques.
(Horowitz et Greenamyre, 2010
) et surtout d’appréhender de possibles interactions avec des contaminants environnementaux comme les pesticides (interactions gène-environnement) (Hatcher et coll., 2008
).
Mécanismes d’action de gènes identifiés dans les formes monogéniques
Le gène
SNCA, premier gène identifié en 1997 dans les formes monogéniques de la maladie de Parkinson et codant pour l’alpha-synucléine, a éveillé toutes les attentions du fait de la présence abondante de son produit dans les corps de Lewy. Les mutations du gène sont nombreuses et peuvent conduire soit à une augmentation de l’expression de la protéine, soit à un dysfonctionnement favorisant son agrégation. En plus du vieillissement qui favorise l’accumulation de l’alpha-synucléine, l’augmentation de la quantité de la protéine peut être liée à une expression plus élevée du transcript et de la protéine (duplications ou triplements du locus du gène, polymorphismes du promoteur ou de la partie 3’ non traduite de l’ARN messager) (Chiba-Falek et Nussbaum, 2001
; Miller et coll., 2004
; Grundemann et coll., 2008
). L’accumulation de la protéine peut aussi être observée en cas d’anomalies de sa voie de dégradation, notamment du système protéasome-ubiquitine
5
Une des raisons du dysfonctionnement de ce système pourrait être un déficit en ATP utilisé par le protéasome, ce qui est logique dans le cas d’une exposition aux pesticides qui perturbent la fonction mitochondriale.
. Toutefois, les modèles animaux (souris transgéniques) favorisant l’accumulation de l’alpha-synucléine évoluent rarement vers un syndrome parkinsonien (défini par la perte des cellules de la substance noire) (Betarbet et coll., 2002
; Di Monte et coll., 2002
). Le lien de cause à effet entre accumulation d’alpha-synucléine et maladie de Parkinson reste donc à préciser. Les mutations présentes sur la protéine alpha-synucléine semblent favoriser son agrégation spontanée. Elles pourraient également réduire le nombre de vésicules dopaminergiques dans les neurones, favorisant ainsi la présence de dopamine cytoplasmique et donc un stress oxydant (Wood-Kaczmar et coll., 2006
). Certains métaux et/ou pesticides stimulent l’agrégation probablement par une augmentation du stress oxydant. Ceci est également vrai pour la dopamine qui forme un adduit sur la protéine (Lundvig et coll., 2005
). La nature hydrophobe ou hydrophile du pesticide pourrait influencer le changement conformationnel à l’origine de l’agrégation (Uversky et coll., 2001
et 2002
; Uversky 2004
; Uversky et Eliezer 2009
).
Une autre protéine impliquée est la LRRK2 (
Leucine-Rich Repeat Kinase 2), une sérine/thréonine kinase et GTPase présente probablement dans un complexe multi-protéique associé aux réseaux de membranes intracellulaires (réticulum, endosomes et mitochondries). Ses différentes mutations sont associées à des manifestations pathologiques variées. À titre d’exemple, la mutation la plus commune (G2019S
6
Une caractéristique intéressante de LRRK2 est que la mutation G2019S a une pénétrance réduite ce qui est en faveur soit d’une interaction avec d’autres gènes ou avec des facteurs de l’environnement.
) augmente l’activité kinase et donc induit une hyper-phosphorylation de ses substrats tels que LRKK2 elle-même (autophosphorylation), la moesine et eIF4E-BP (
eukaryotic Initiation Factor 4E-binding protein) (Horowitz et Greenamyre, 2010
). Les souris invalidées pour la LRRK2 ne semblent pas présenter d’anomalies et se développent normalement jusqu’à l’âge adulte (Andres-Mateos et coll., 2009
). L’absence de LRRK2 ne rend pas les souris plus sensibles au développement d’une maladie de Parkinson induite par le MPTP (voir ci-dessous) ce qui laisse sous-entendre que le lien avec la maladie n’est pas dû à une perte de fonction mais bel et bien à un gain (Andres-Mateos et coll., 2009
). Chez le nématode (
Caenorhabditis elegans), la surexpression de LRRK2 a un effet protecteur vis-à-vis d’un pesticide, le paraquat (Saha et coll., 2009
).
La parkine, une E3-ubiquitine ligase, a été également impliquée. Les mutations de cette protéine sont associées à une forme récessive de parkinsonisme d’évolution lente (des polymorphismes sont retrouvés dans certains cas sporadiques). De façon surprenante, l’analyse histopathologique ne retrouve pas toujours des corps de Lewy, et une accumulation de tous les substrats de la protéine. Par ailleurs, l’invalidation de ce gène chez la souris entraîne un défaut de fonctionnement mitochondrial et un stress oxydant sans perte de neurones dopaminergiques (Palacino et coll., 2004
; Horowitz et Greenamyre, 2010
). Ces données suggèrent que la fonction de la parkine dans la dégradation des protéines n’est pas celle impliquée dans la pathologie. Des articles récents suggèrent que la parkine serait spécifiquement recrutée par la protéine Pink1 (
PTEN-induced putative kinase 1, une kinase mitochondriale stabilisée par la dépolarisation) au niveau de mitochondries dépolarisées (dont le fonctionnement est anormal). Elle stimulerait leur élimination par mitophagie (autophagie mitochondriale) après ubiquitinylation d’un canal membranaire voltage-dépendant (VDAC1) et recrutement d’un adaptateur p62, responsable du ciblage de protéines ubiquitinylées aux vacuoles autophagiques (Narendra et coll., 2010
; Narendra et Youle, 2011
). Des mutations de Pink1 sont retrouvées dans des formes monogéniques de maladie de Parkinson. Le rôle sensibilisateur des mutations de la parkine ou de Pink1 vis-à-vis d’une exposition à des pesticides (roténone, MPTP) a également été démontré (Palacino et coll., 2004
; Casarejos et coll., 2006
; Wang et coll., 2007
; Haque et coll., 2008
).
La protéine DJ-1, exprimée majoritairement dans les astrocytes, est sensible au stress oxydant qui provoque sa translocation du cytoplasme vers la mitochondrie où elle exercerait une fonction protectrice mal comprise (Canet-Aviles et coll., 2004
). Des mutations ont été décrites pour cette protéine dans de rares cas de parkinsonisme récessif précoce. La perte de fonction de DJ-1 pourrait conduire à une surexpression du transporteur de la dopamine dans le striatum conférant aux neurones dopaminergiques, une plus grande capacité d’accumulation de composés toxiques.
Le fait que sur les cinq protéines identifiées, trois soient exprimées dans la mitochondrie, suggère que la mitochondrie joue un rôle central dans les mécanismes pathologiques de la maladie de Parkinson.
Interactions gène-environnement : un rôle des pesticides ?
Une interaction forte entre gènes et environnement a été suspectée du fait par exemple, d’une pénétrance incomplète de certaines formes monogéniques (Horowitz et Greenamyre, 2010
). Plusieurs facteurs non génétiques ont été associés avec la maladie de Parkinson (Horowitz et Greenamyre, 2010
) : l’âge, le genre (mâle), le tabagisme (association inverse avec un rôle possible de la nicotine dans la protection spécifique des neurones dopaminergiques, Quik et coll., 2008
), l’exposition à la caféine (association inverse) (Ascherio et coll., 2001
; Cersosimo et Koller, 2006
; Elbaz et Moisan, 2008
), ou les traumatismes crâniens (Bower et coll., 2003
; Rugbjerg et coll., 2008
). Certaines conditions physiopathologiques (diabète de type 2, obésité) sont suspectées de favoriser l’apparition d’une maladie de Parkinson (Horowitz et Greenamyre, 2010
). Les niveaux plasmatiques d’urate sont inversement corrélés avec le risque de maladie de Parkinson (Weisskopf et coll., 2007
).
Maladie de Parkinson : lien avec les pesticides
Historiquement, les premières suspicions d’un lien entre pesticides et maladie de Parkinson découlent de l’analyse d’une exposition accidentelle (Langston Ballard et coll., 1983
). De jeunes toxicomanes californiens synthétisèrent et consommèrent involontairement une neurotoxine, la 1-méthyl-4-phényl-1,2,3,6-tetrahydropyridine (ou MPTP) avec pour conséquence, la survenue précoce d’un syndrome parkinsonien amélioré par le traitement par la levodopa.
MPTP
Nature chimique de la molécule, voies d’entrée, métabolisme, passage de la barrière hémato-encéphalique
Le MPTP présente une forte analogie structurale avec un pesticide, le paraquat (figure 23.3
).
La MPTP traverse facilement la barrière hémato-encéphalique (BHE), puis elle est métabolisée dans les astrocytes par la monoamine oxydase B (MAO-B) en MPP+ (1-méthyl-4-phenylpyridinium) qui est capté par le transporteur de la dopamine (DAT) des cellules neuronales et provoque la mort sélective de ces neurones.
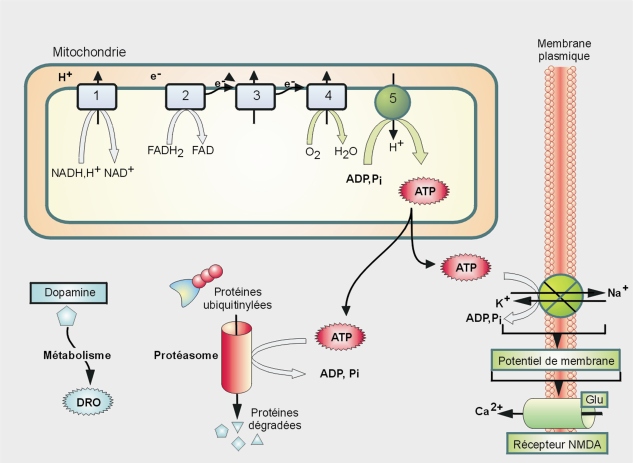
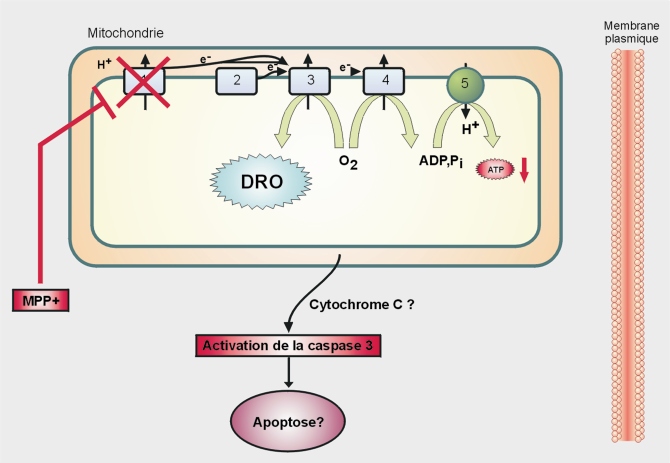
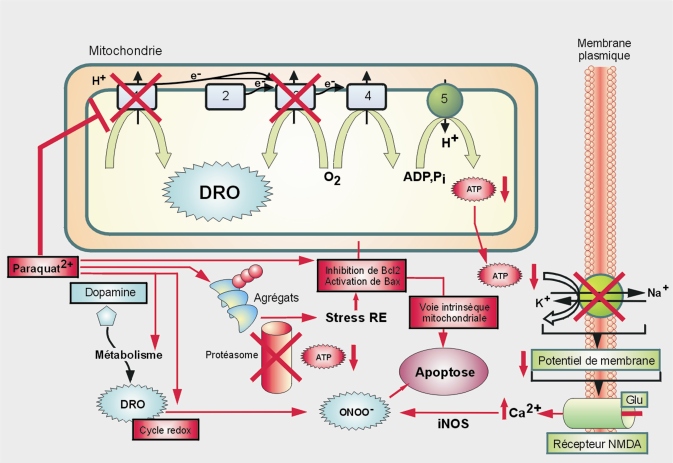
Mécanisme d’action
Le MPP+ cible les mitochondries, en inhibant notamment le complexe I de la chaîne respiratoire (figure 23.4
) (Nicklas et coll., 1992
; Greenamyre et coll., 1999
et 2001
; Dauer et coll., 2002
; Sherer et coll., 2002
). Ceci a pour conséquence :
• une diminution de la production d’ATP, essentiel pour la cellule (traduction, maintenance du potentiel membranaire…) ;
• une fuite d’électrons par la chaîne de transport vers l’O
2 avec pour conséquence, la production de dérivés réactifs de l’oxygène (DRO) (figure 23.4
).
Ces données, quant aux effets du MPP+ sur la chaîne respiratoire, ont conduit à des travaux portant sur la fonctionnalité des mitochondries ou sur le niveau de stress oxydant soit dans des échantillons de patients, soit sur modèles animaux mimant la maladie de Parkinson. Une diminution de 30 % de l’activité du complexe I a été montrée dans différents tissus de malades (Foley et Riederer, 2000
). Ces effets du MPP+ ont suscité de très nombreux travaux de recherche sur l’action des pesticides sur la fonction mitochondriale et le stress oxydant ainsi que sur les voies de signalisation qu’ils régulent comme l’apoptose. L’implication de l’apoptose dans la neurodégénérescence des neurones de la substance noire exposés au MPTP a été démontrée
in vivo (par des mesures de l’activation de la caspase 3) (Hartmann et coll., 2000
) et
in vitro (par un blocage de l’apoptose de cellules de neuroblastome SH-EP1 par un traitement à l’
Insulin-like Growth Factor 1 ou IGF-1) (Wang et coll., 2010
).
Concernant l’interaction gène-environnement, le MPTP ne favorise pas la dégénération dopaminergique chez les animaux parkine -/- par rapport aux témoins (Palacino, 2004
). En revanche, dans des neurones déficients en Pink1, la réexpression d’une forme normale de Pink1, protège ceux-ci vis-à-vis du MPP+ (Haque et coll., 2008
). Les mutations de DJ-1 dans différents modèles animaux (souris, drosophile) montrent une sensibilisation des neurones à certains pesticides ou dérivés comme le MPTP (Kim et coll., 2005
; Menzies, et coll., 2005
; Meulener et coll., 2005
; Yang et Tiffany-Castiglioni, 2005
; Lavara-Culebras et Paricio, 2007
).
Paraquat (PQ) et diquat (herbicides)
Comme le paraquat présente une grande analogie structurale avec le MPP+, ceci a conduit à de nombreuses études portant sur le lien éventuel entre exposition à ce pesticide et survenue de symptômes parkinsoniens.
Nature chimique de la molécule, voies d’entrée, métabolisme, passage de la barrière hémato-encéphalique
Le paraquat (PQ ou chlorure de 1,1’-dimethyl-4,4’-bipyridinium, figure 23.5
) est un herbicide non sélectif très utilisé en agriculture depuis 1961, extrêmement toxique pour le système pulmonaire, faiblement métabolisé et excrété dans les urines, quasiment inchangé. La flore intestinale peut métaboliser le PQ par déméthylation ou oxydation (Murray et Gibson, 1974
; Shimada et coll., 2002
).
Le paraquat ne traverse pas passivement les membranes lipidiques. Les premières études sur la biodistribution du PQ en particulier dans le système nerveux central, menées dans les années 1990, indiquaient une diffusion du PQ dans le cerveau au niveau des zones sans barrière hémato-encéphalique (BHE) comme l’hypothalamus ou situées à l’extérieur de la BHE (épiphyse) (Naylor et coll., 1995
; Widdowson et coll., 1996 a
et b
). Les données les plus récentes modulent cette constatation en démontrant que le PQ atteint bien les zones du cerveau jouant un rôle déterminant dans la maladie de Parkinson (avec une demi-vie de 4 semaines après une seule injection à la concentration de 10 mg/kg) (Corasaniti et coll., 1998
; Prasad et coll., 2007
). Celui-ci tend même à s’accumuler après plusieurs administrations. L’une de ces études (impliquant des rats sous microdialyse, une analyse HPLC/UV et différentes doses de PQ) a permis de proposer un mécanisme de transport : l’accumulation de PQ dans le cerveau est probablement saturable (sans perte d’intégrité de la BHE) et impliquerait un système de transport actif dépendant du sodium, responsable par ailleurs du transport des acides aminés neutres (comme la leucine, capable d’inhiber le transport) (Shimizu et coll., 2001
; McCormack et Di Monte 2003
; Chanyachukul et coll., 2004
). L’utilisation d’inhibiteurs suggère aussi l’implication d’un transporteur de dopamine (Shimizu et coll., 2003
). Cette donnée permettrait de comprendre la spécificité de la toxicité du PQ vis-à-vis des neurones dopaminergiques (qui expriment ces transporteurs dopaminergiques permettant la recapture du neurotransmetteur). Dans la lignée de ces travaux, plusieurs études ont montré que le blocage du transporteur classique de la dopamine (DAT) les protège des effets du PQ (McCormack et Di Monte, 2003
; Shimizu et coll., 2003
; Yang et Tiffany-Castiglioni, 2005
). Cette influence du DAT est toutefois remise en question par certaines études (Richardson et coll., 2005
).
Par ailleurs, la toxicocinétique du PQ dans le système nerveux central semble particulière par rapport aux autres organes : l’élimination de la molécule par le cerveau semble être beaucoup plus lente (Prasad et coll., 2007
; Prasad et coll., 2009
). De plus, la concentration de PQ augmente de 2 à 3 fois dans plusieurs régions du cerveau si une co-exposition est réalisée avec du manèbe (Barlow et coll., 2003
). Il est possible que ce fongicide modifie les niveaux d’expression ou l’activité de transporteurs du PQ. Il faut souligner qu’une controverse existe quant au passage de la BHE par le PQ avec un certain nombre d’études mentionnant que celui-ci ne traverse pas cette barrière (Miller et coll., 1999
; Barlow et coll. 2003
; Bartlett et coll., 2009
et 2011
).
Mécanismes d’action
Les premières études
in vivo menées avec le PQ, montrent sa neurotoxicité pour toutes les zones du cerveau au niveau desquelles il est injecté, sans spécificité (vis-à-vis par exemple de la substance noire). En revanche, des études utilisant une administration systémique de PQ montrent une spécificité de ses cibles avec la perte des neurones dopaminergiques, dépendante de l’âge de l’animal et de la dose (Brooks et coll., 1999
; Prasad et coll., 2009
), quand les autres populations neuronales de la substance noire ne sont pas atteintes (neurones GABAergiques de la
pars reticulata), de même l’hippocampe est épargné, ce qui souligne entre autres la sensibilité des neurones dopaminergiques (Manning-Bog et coll., 2002
; McCormack et coll., 2002
). Des études histologiques (immunohistochimie de la tyrosine hydroxylase qui intervient dans la synthèse de la L-dopa) confirment d’ailleurs l’existence de populations neuronales plus ou moins sensibles au sein de la substance noire (Ossowska et coll., 2005a
et b
).
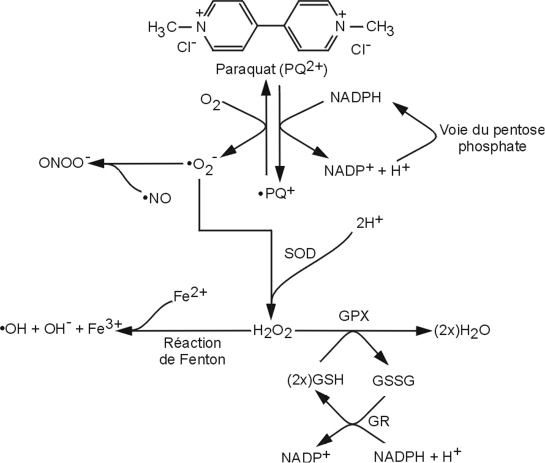
Concernant le stress oxydant et les mitochondries, le PQ exerce ses effets toxiques par la production de radicaux libres, entretenue par un cycle d’oxydoréduction. Au cours de celui-ci (figure 23.5
), l’ion PQ
2+ capte un électron d’un réducteur (il peut s’agir soit du NADPH,H
+, soit d’électrons de la chaîne respiratoire mitochondriale) et le transfère à l’O
2 produisant l’anion superoxyde O
2-° ce qui permet la régénération de l’ion PQ
2+. La présence de PQ dans une cellule permet ainsi une production continue d’O
2-° et de ses dérivés (peroxyde d’hydrogène, H
2O
2 et radical superoxyde, OH°) (Moretto et Colosio, 2011
).
Le niveau d’expression des enzymes éliminant ces dérivés réactifs de l’oxygène ou DRO (par exemple : la superoxyde dismutase ou SOD), est donc un paramètre clé conditionnant la sensibilité des cellules à l’action oxydante du PQ (Iannone et coll., 1991
; Rossi et coll., 2001
; Thiruchelvam et coll., 2005
; Choi et coll., 2006
). Le rôle d’enzymes du métabolisme des xénobiotiques pourrait être fondamental (Ghersi-Egea et coll., 1991
; Dutheil et coll., 2008
) tout comme celui de facteurs de transcription comme Nrf2 (
NF-E2-related factor-2), activé par le stress oxydant et favorisant l’expression de l’hème oxygénase 1 (HO1), impliquée dans la dégradation de l’hème et empêchant la production de OH° à partir de H
2O
2 (Minelli et coll., 2009
).
Après transport par une protéine dépendante de la polarité membranaire mitochondriale, le PQ exercerait ses effets soit au niveau du complexe 1 (Tawara et coll., 1996
; Cocheme et Murphy, 2008
), soit au niveau du complexe 3 (Castello et coll., 2007
; Drechsel et Patel, 2009
) (figure 23.6
). L’inhibition du complexe 1 est toutefois controversée (Choi et coll., 2008
). Chez le nématode (
C. elegans), la surexpression de LRRK2, localisée au niveau des membranes dont celles de la mitochondrie, protège des effets neurotoxiques du PQ (Saha et coll., 2009
). La compréhension de la fonction LRRK2 représente donc une piste intéressante.
En plus du cycle d’oxydoréduction et des effets sur la chaîne respiratoire, le PQ pourrait augmenter l’expression de la NADPH oxydase (Wu et coll., 2005
; Miller et coll., 2007
; Purisai et coll., 2007
; Cristovao et coll., 2009
) et de la xanthine oxydase (études menées sur des cellules granulaires de cervelet), toutes deux productrices de DRO (Gonzalez-Polo et coll., 2004
). Une étude de Kang et coll. (2009
) rapporte par ailleurs que le PQ stimulerait le métabolisme oxydatif de la dopamine et diminuerait également les stocks de glutathion (Schmuck et coll., 2002
; Osakada et coll., 2003
et 2004
; Kang et coll., 2009
). Dans la lignée de ces travaux, l’ajout de dopamine à des cultures cellulaires favorise la toxicité du PQ mais pas celle d’autres pesticides (Fei et Ethell, 2008
). L’effet du PQ sur le métabolisme de la dopamine expliquerait donc pourquoi les neurones dopaminergiques seraient plus sensibles au stress oxydant (McCormack et coll., 2002
et 2006
; Richardson et coll., 2005
). Le niveau de peroxydation lipidique mesuré après traitement par le PQ chez des souris, est significativement plus élevé dans la substance noire que dans les autres parties du cerveau (Tawara et coll., 1996
; Prasad et coll., 2007
).
Concernant la formation d’agrégats, en plus de son effet sur la production de DRO, le PQ est suspecté d’activer la formation des corps de Lewy contenant de l’alpha-synucléine (et/ou de la parkine) (Uversky et coll., 2001
; Manning-Bog et coll., 2002
; Thiruchelvam et coll., 2004
; Wang et coll., 2005
; Fernagut et coll., 2007
; Norris et coll., 2007
). Cet effet semble toutefois réversible (Manning-Bog et coll., 2002
) et pourrait en fait correspondre à une réponse protectrice vis-à-vis du PQ (Manning-Bog et coll., 2003
). Une étude récente montre que d’autres protéines sont susceptibles de s’agréger en présence de PQ comme la pompe calcique neuronale (PMCA, plasma membrane Ca
2+ - ATPase) (Zaidi et coll., 2009
).
Concernant l’excitotoxicité, l’administration de PQ au niveau du striatum de rats (par injection spécifique) conduit à une élévation transitoire des niveaux extracellulaires de glutamate, et durable des niveaux de dopamine (Shimizu et coll., 2003
). Ceci pourrait contribuer à un mécanisme d’excitotoxicité passant par une stimulation séquentielle de récepteurs glutamatergiques (non-NMDA et NMDA), qui conduirait à une entrée de Ca
2+ au niveau des terminaisons nerveuses dopaminergiques et à une activation de la NO synthase inductible (iNOS). Le PQ en stimulant à la fois la production de DRO (voir ci-dessus) et celle de NO par la iNOS, serait responsable de l’augmentation des niveaux de peroxynitrite (ONOO
-) (produit d’une réaction spontanée entre NO° et O
2-°) qui réagit fortement avec l’ADN et les lipides, conduisant ainsi à la mort cellulaire. Ce rôle du glutamate avait déjà été suggéré en 1992 par l’effet protecteur d’un antagoniste du récepteur NMDA (MK801) vis-à-vis du PQ (neurotoxicité dans l’hippocampe) (Bagetta et coll., 1992
).
Le rôle des cellules gliales
7
Cellules entourant les neurones et assurant diverses fonctions vis‑à‑vis de ceux‑ci (protection, nutrition…)
dans la toxicité du PQ et du diquat
8
Le diquat pourrait lui aussi exercer des effets pro‑oxydants soit au sein des neurones dopaminergiques soit au sein des cellules gliales. Son exposition a été aussi associée à un risque de maladie de Parkinson.
pourrait être non négligeable. En réponse au PQ, les cellules gliales expriment à la membrane la protéine Fas-L (ligand) qui lie le récepteur Fas présent sur les cellules neuronales dopaminergiques, activant ainsi la voie apoptotique extrinsèque (et certaines caspases spécifiques) (Vogt et coll., 1998
). Par ailleurs, une étude
in vitro montre que la toxicité du PQ à de faibles concentrations ne s’observe qu’en présence de cellules gliales génératrices d’un stress oxydant produit par la NADPH oxydase selon un mécanisme impliquant les MAP kinases ERK (
Extracellular Regulated Kinase) et PKC (
Protein Kinase C) (Wu et coll., 2005
; Purisai et coll., 2007
; Kim et coll., 2008
). Ce rôle activateur de la glie est évoqué par les études conduites avec le mélange PQ et manèbe (voir dans le paragraphe consacré au manèbe).
La mort cellulaire sélective des neurones dopaminergiques pourrait être liée à l’ensemble des phénomènes décrits précédemment. La nécrose est observée à hautes doses
in vivo (Calo et coll., 1990
). Par ailleurs, les neurones de sujets parkinsoniens montrent des signes d’autophagie et le PQ semble activer certaines voies de signalisation autophagiques (Gonzalez-Polo et coll., 2007
et 2009
). Toutefois, le plus grand nombre de publications portant sur le rôle du PQ dans la mort cellulaire, porte sur l’apoptose (Li et Sun, 1999
) : le PQ semble activer principalement la voie intrinsèque mitochondriale
9
Une étude suggère toutefois que la voie extrinsèque pourrait aussi être impliquée (Vogt et coll., 1998
).
par stimulation des protéines pro-apoptotiques Bax, Bak, Bid et Noxa qui favorisent la libération mitochondriale de cytochrome C et l’activation des caspases 3 et 9 (Gonzalez-Polo et coll., 2004
; Yang et Tiffany-Castiglioni 2005
; Fei et Ethell 2008
). À l’inverse, les protéines anti-apoptotiques comme Bcl2 semblent exercer un effet protecteur comme le suggère une étude menée sur des souris déficientes en Bcl2 traitées avec du PQ, plus sensibles que des souris sauvages (Hochman et coll., 1998
). Par ailleurs, le PQ active la protéine p53 et deux de ses cibles transcriptionnelles : Bax et Noxa (Rossi et coll., 2001
; Yang et Tiffany-Castiglioni 2008
). Un autre mécanisme d’activation de Bax pourrait impliquer les agrégats protéiques. En effet, l’accumulation de protéines mal conformées est susceptible de provoquer un stress du réticulum endoplasmique à l’origine d’une activation des SAPK/JNK (
Stress-Activated Protein Kinases/
c-Jun-N-terminal kinases) et de la protéine CHOP (
C/EBP Homologous Protein), un inhibiteur de Bcl2 (Chun et coll., 2001
; Peng et coll., 2004
; Niso-Santano et coll., 2006
; Klintworth et coll., 2007
; Ramachandiran et coll., 2007
; Yang et coll., 2009
; Niso-Santano et coll., 2010
). L’inhibition de Bcl2 conduit à une activation de Bax. Les SAPK pourraient être activées indirectement par les DRO (redox cycle du PQ) (Ramachandiran et coll., 2007
; Yang et coll., 2009
; Niso-Santano et coll., 2010
). Les inhibiteurs des SAPK/JNK pourraient représenter des agents thérapeutiques potentiels dans la maladie de Parkinson. Malgré les similitudes structurales évoquées précédemment, l’activation de cette voie ne semble pas s’accomplir avec le MPTP ou la roténone (voir plus loin).
En résumé, l’impact du paraquat (hydrophile) sur le SNC suppose le franchissement de la barrière hématoencéphalique. Certains travaux suggèrent un système de transport actif dépendant du sodium. Les niveaux d’expression ou d’activité de transporteurs du PQ pourrait être modulés par une co-exposition avec du manèbe qui augmente la concentration de PQ dans plusieurs régions du cerveau. La neurotoxicité du PQ affecte plus spécifiquement les neurones dopaminergiques. Le PQ exerce ses effets toxiques par la production de dérivés réactifs de l’oxygène (DRO). La mitochondrie semble jouer un rôle majeur dans la progression de la pathologie tant du point de vue de la production de DRO que de l’activation de l’apoptose, son dysfonctionnement expliquant par ailleurs potentiellement d’autres phénomènes (déplétion en ATP, inhibition du protéasome, agrégation protéique). Un rôle activateur de la glie est évoqué. L’ensemble de ces éléments suggère une plausibilité biologique avec la maladie de Parkinson.
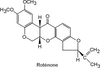
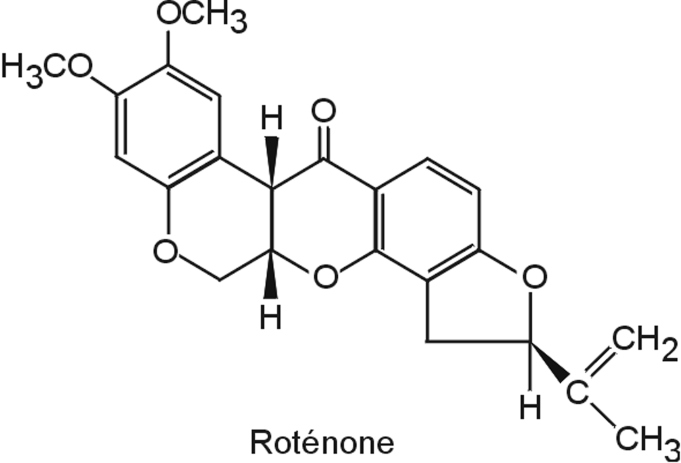
Roténone
Nature de la molécule chimique, métabolisme, passage de la barrière hémato-encéphalique
La roténone (figure 23.7
) est un pesticide dérivé naturel de plantes utilisé pour limiter la prolifération de populations de poissons indésirables et en agriculture biologique.
Son usage limité l’empêche probablement d’être un contributeur majeur du développement de la maladie de Parkinson mais son mécanisme d’action assez bien caractérisé a permis de mieux comprendre certains aspects mécanistiques associés à la neurodégénérescence des neurones dopaminergiques. La roténone, extrêmement hydrophobe, diffuse facilement à travers les membranes cellulaires. Cette propriété explique pourquoi elle atteint facilement les cellules du système nerveux central sans l’intervention d’un système de transport.
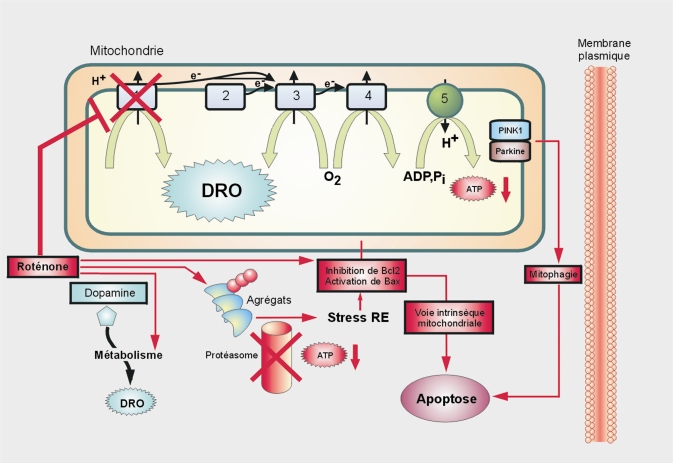
Mécanismes d’action
L’analyse du métabolisme de la roténone montre que celle-ci est responsable des effets et non ses métabolites (Caboni et coll., 2004
). L’administration subchronique (voie intraveineuse de doses de 2-3 mg/kg/jour pendant 36 jours administrées à l’aide d’une mini-pompe) de roténone à des rats conduit à un syndrome parkinsonien (avec des manifestations neurologiques et non neurologiques). Les terminaisons nerveuses striatales et la substance noire sont atteintes chez 12 animaux traités sur 25 (Betarbet et coll., 2000
). D’autres études menées sur d’autres modèles murins montrent dans certains cas, une atteinte d’autres parties du cerveau (striatum, globlus pallidus) mais sans affecter la substance noire (Thiffault et coll., 2000
) ou les corps cellulaires des neurones dopaminergiques (Ferrante et coll., 1997
) tandis que dans d’autres cas, une importante déplétion de dopamine est retrouvée au niveau de la substance noire (Alam et Schmidt, 2002
). Ceci suggère que la sensibilité du modèle dépend de la structure et de l’organisation cérébrale de chaque animal mais aussi du protocole utilisé. Le contexte génétique joue un rôle important. Ainsi, des cellules neuronales de souris invalidées pour la parkine (Casarejos et coll., 2006
) ou des souches de drosophile portant une mutation de la parkine (Parkine R275W), présentent une plus grande sensibilité vis-à-vis de la roténone (Wang et coll., 2007
).
Concernant le stress oxydant, le dysfonctionnement mitochondrial, la formation d’agrégats et le rôle des cellules gliales, sur le plan histopathologique ou mécanistique, la roténone provoque l’accumulation et l’agrégation d’alpha-synucléine (et l’apparition de structures d’inclusion ressemblant à des précurseurs de corps de Lewy) ainsi que la translocation mitochondriale de DJ-1 (Sherer et coll., 2003
). L’accumulation de certains agrégats protéiques pourrait être due à une inhibition du protéasome. Toutefois, la roténone est plus particulièrement connue pour inhiber le complexe 1 de la chaîne respiratoire (figure 23.8
) (Seaton et coll., 1997
). Cette inhibition conduit à la fois à une baisse importante de la production d’ATP et à la synthèse de DRO et/ou de NO°, potentiellement responsables de la mort cellulaire (He et coll., 2003
; Sherer et coll., 2003
; Bashkatova et coll., 2004
). Toutefois, dans l’étude de Betarbet et coll. (2000
), sur les effets sub-chroniques de la roténone, l’inhibition du complexe 1 aux doses provoquant le syndrome parkinsonien, ne semble pas suffisante pour bloquer significativement la phosphorylation oxydative et la production d’ATP. Un autre mécanisme pourrait impliquer la parkine : des travaux récents suggèrent en effet qu’une dépolarisation mitochondriale (obtenue avec la roténone) favorise la stabilisation (par inactivation d’une protéase mitochondriale, dépendante du potentiel de membrane) de la protéine Pink1 (
PTEN-induced putative kinase 1), une kinase mitochondriale dont les mutations (domaine kinase) ont été retrouvées dans des formes monogéniques de maladie de Parkinson, Pink1 recrute alors la parkine. Le bon fonctionnement du domaine kinase de Pink1 semble essentiel à cette interaction (mais sans phosphorylation de la parkine). Ce mécanisme stimulerait l’élimination des mitochondries dépolarisées par mitophagie (autophagie mitochondriale) après ubiquitinylation d’un canal membranaire voltage-dépendant (VDAC1) et recrutement d’un adaptateur p62, responsable du ciblage de protéines ubiquitinylées aux vacuoles autophagiques (Narendra et coll., 2008
et 2010
). Cette étude pourrait donc rendre compte d’une interaction potentielle entre gènes et environnement en cas de déficit de fonction de la parkine et exposition à certains pesticides qui ciblent la mitochondrie.
Les études
in vitro menées sur des lignées cellulaires ou des cultures primaires de neurones (ou co-cultures avec des cellules gliales), montrent que la roténone (à des concentrations basses de l’ordre du nanomolaire) augmente l’expression de l’alpha-synucléine et provoque un stress oxydant à l’origine d’une baisse des concentrations de glutathion et d’une activation de certaines voies apoptotiques (Gao et coll., 2002
et 2003
; Sherer et coll., 2002
; Sugeno et coll., 2008
). Gao et coll. montrent notamment qu’à des concentrations très faibles (<1 nM, 8 jours), la roténone induit une neurodégénérescence sélective des cellules dopaminergiques dans des co-cultures de neurones et de cellules gliales. La présence des cellules gliales augmente considérablement cette sensibilité. Ce phénomène est retrouvé en présence de lipopolysaccharide (LPS), une molécule pro-inflammatoire en l’absence de cellules gliales (Ling et coll., 2004
). Le rôle des cellules gliales a été décortiqué par plusieurs études : la NADPH oxydase gliale par la production de DRO, pourrait ainsi sensibiliser les neurones dopaminergiques à l’action de la roténone (Liu et coll., 2003
) et intervenir en préambule des mécanismes neurodégénératifs (Sherer et coll., 2003
; Shaikh et Nicholson, 2009
). Enfin, des études récentes montrent que la roténone inhibe la redistribution vésiculaire de dopamine, favorisant son accumulation cytoplasmique (et potentiellement un stress oxydant) (Ahmadi et coll., 2008
; Watabe et Nakaki, 2008
).
Concernant la mort cellulaire, une étude est parvenue à reproduire des caractéristiques de maladie de Parkinson chez la Drosophile exposée à la roténone (Coulom et Birman, 2004
). L’apoptose activée par la roténone pourrait être liée à la voie intrinsèque mitochondriale (Ahmadi et coll., 2003
; Pei et coll., 2003
; Watabe et Nakaki 2004
; Clayton et coll., 2005
; Radad et coll., 2006
; Lee et coll., 2008
). La polarisation membranaire mitochondriale pourrait ainsi jouer un rôle comme le suggère l’effet protecteur d’une activation de canaux potassiques mitochondriaux sensibles à l’ATP (Tai et Truong 2002
; Tai et coll., 2003
). Deux études suggèrent toutefois que des mécanismes indépendants des caspases pourraient intervenir (Li et coll., 2005
; Lim et coll., 2007
). D’autres signaux pro-apoptotiques pourraient être activés par la roténone comme certaines kinases (par exemple JNK) ou le stress du réticulum endoplasmique (Ryu et coll., 2002
; Newhouse et coll., 2004
; King et Jope, 2005
; Gimenez-Cassina et coll., 2009
). Le rôle du contexte génétique semble non négligeable car la roténone favorise la neurodégénérescence dans un contexte de surexpression de l’alpha-synucléine ou de réduction des niveaux de parkine, DJ-1 ou pink1 (Deng et coll., 2005
; Ved et coll., 2005
; Casarejos et coll., 2006
). Toutefois, bien que des analogies (structurales et mécanistiques) existent entre roténone et PQ/MPTP, leur mode d’action ne semble pas complètement équivalent comme le suggèrent plusieurs études (Bové et coll., 2005 ; Richardson et coll., 2005
; Ramachandiran et coll., 2007
).
En résumé, la roténone, composé naturel hydrophobe, diffuse facilement à travers les membranes cellulaires. In vitro, on observe une neurodégénérescence sélective des cellules dopaminergiques dans des co-cultures de neurones et de cellules gliales. De faibles concentrations augmentent l’expression de l’alpha-synucléine et provoquent un stress oxydant à l’origine d’une baisse des concentrations de glutathion et d’une activation de certaines voies apoptotiques. La présence des cellules gliales augmente considérablement cette sensibilité. L’ensemble des éléments suggère une plausibilité biologique avec la maladie de Parkinson.
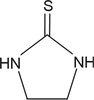
Dithiocarbamates (mancozèbe, manèbe, thiram, zirame)
Introduction sur la nature chimique de la molécule et métabolisme
Les dithiocarbamates sont des fongicides-carbamates, n’inhibant pas l’acétylcholine estérase. Ils forment des complexes lipophiles avec des cations di- ou trivalents. Quatre classes de dithiocarbamates existent parmi lesquels les éthylène-bis-dithiocarbamates (EBDC) dont le manèbe qui comporte à la fois une partie métallique (manganèse, potentiellement toxique) et une partie organique. Le produit de dégradation des EBDC, l’éthylènethiourée (ETU, figure 23.9
), a été impliqué comme perturbateur de plusieurs processus cellulaires dont la synthèse des hormones thyroïdiennes (Doerge et Takazawa 1990
; Marinovich et coll., 1997
).
Certaines études associent l’exposition aux dithiocarbamates à des syndromes parkinsoniens (Ferraz et coll., 1988
; Hoogenraad, 1988
; Meco et coll., 1994
).
Mécanismes d’action
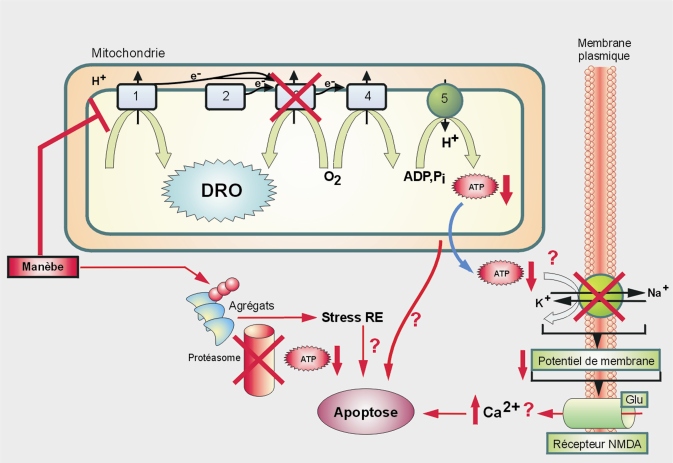
Comme le PQ et la roténone, les dithiocarbamates pourraient conduire à la mort des neurones dopaminergiques via plusieurs processus dérégulés (figure 23.10
). Cependant, il est difficile d’identifier un mécanisme d’action commun à tous les dithiocarbamates. Chez des modèles animaux, l’exposition au manèbe conduit à une diminution de l’activité locomotrice (et une perturbation de la réponse à d’autres tests neurocomportementaux) (Ferraz et coll., 1988
). L’exposition à de faibles concentrations de manèbe (500 nM) pendant quelques minutes peut favoriser
in vitro l’augmentation de la concentration de dopamine et la production de DRO (Barlow et coll., 2003
). Des études ont démontré une inhibition directe du complexe 3 de la chaîne respiratoire mitochondriale (Zhang et coll., 2003
; Domico et coll., 2006
). Le manèbe induit par ailleurs l’auto-oxydation de dérivés métaboliques de la dopamine (Fitsanakis et coll., 2002
), diminue les niveaux intracellulaires de glutathion (Barlow et coll., 2005
) et inhibe la fonction du protéasome favorisant par exemple, l’agrégation de l’alpha-synucléine (Zhou et coll., 2004
). Cet effet sur le protéasome est retrouvé avec un autre dithiocarbamate, le zirame (Chou et coll., 2008
). L’apoptose de cellules dopaminergiques (PC12) a été observée en présence de plusieurs dithiocarbamates, par un mécanisme impliquant une augmentation de l’influx calcique (Sook Han et coll., 2003
).
En résumé, il est difficile d’identifier un mécanisme d’action commun à tous les dithiocarbamates. Les dithiocarbamates (manèbe) pourraient conduire à la mort des neurones dopaminergiques via plusieurs processus dérégulés. Associés en mélange, ils pourraient accentuer la toxicité d’autres pesticides. Les mécanismes évoqués pour le manèbe en particulier semblent possiblement impliqués dans la maladie de Parkinson.
Effets combinés du manèbe et d’autres pesticides
Les effets combinés du manèbe et d’autres pesticides (PQ, Roténone, MPP+)
10
Le manèbe et le paraquat sont parfois utilisés dans les mêmes zones géographiques.
ont été étudiés sur différents modèles (souris, rats, mitochondries isolées) : certaines études montrent un effet plus sévère du mélange (à hautes doses) que les composés individuels (exemple : déficit moteur associé à une atteinte de la recapture de la dopamine au niveau striatal) (Takahashi et coll., 1989
; Bachurin et coll., 1996
; McGrew et coll., 2000
; Thiruchelvam et coll., 2000a
et b
, 2002
et 2003
; Cicchetti et coll., 2005
; Cory-Slechta et coll., 2005
). Par ailleurs, une première exposition chez de jeunes animaux pourrait les sensibiliser en cas d’exposition à l’âge adulte (Cory-Slechta et coll., 2005
). Enfin, le manèbe augmente de 2 à 3 fois la concentration de PQ dans plusieurs régions du cerveau (voir aussi le paragraphe PQ) (Barlow et coll., 2003
).
Sur le plan mécanistique, deux études récentes montrent que le mélange PQ+manèbe, en plus d’affecter les activités locomotrices de souris traitées, diminue les niveaux de dopamine dans le striatum, de tyrosine hydroxylase (qui intervient dans la synthèse de la L-Dopa) et du transporteur vésiculaire des monoamines (VMAT2, suggérant une possibilité de stress oxydant cytoplasmique par baisse de la recapture), augmente la mort cellulaire de neurones de la substance noire et les niveaux de cytochrome P450, CYP2E1 (générateur potentiel de stress oxydant), de glutathion-S-transférase A4-4 (adaptation au stress oxydant), de NO synthase inductible (iNOS) et de caspase 9 (Gupta et coll., 2010
). Cet effet sur la caspase 9 renforce l’idée d’une action pro-apoptotique du mélange : la caractérisation de mécanismes d’action a conduit à l’identification de Bik et Bim comme cible du mélange. Ces protéines sont des activateurs de la voie Bax qui régulent la voie intrinsèque mitochondriale (voir paragraphe PQ) (Thiruchelvam et coll., 2002
et 2003
; Fei et Ethell 2008
). Le mélange est suspecté d’activer les cellules gliales (moins sensibles au stress oxydant) et de provoquer indirectement la mort des cellules neuronales (voir paragraphe PQ, activation de la voie extrinsèque) (Vogt et coll., 1998
; Cicchetti et coll., 2005
; Saint-Pierre et coll., 2006
). Le rôle du stress oxydant dans ces effets semble majeur comme le suggèrent les effets protecteurs du glutathion (Zeevalk et coll., 2010
).
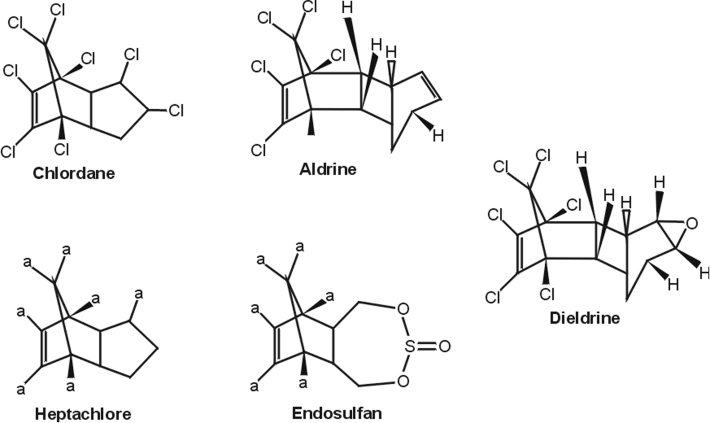
Organochlorés (aldrine, chlordane, DDT, dieldrine, endosulfan, endrine, heptachlore, lindane)
Nature de la molécule chimique, métabolisme, passage de la barrière hémato-encéphalique
Les organochlorés (OC) sont des insecticides divisés en 3 classes. Les dichlorodiphényléthanes (exemple : DTT, DDE) et les hexachlorocyclohexanes (exemple : lindane) n’ont pas été impliqués dans la maladie de Parkinson. Ils peuvent toutefois représenter des marqueurs d’exposition à des mélanges de pesticides. Les principales données établissant un lien avec la maladie de Parkinson, concernent la 3
e classe des cyclodiènes (aldrine, dieldrine, chlordane, endosulfan, heptachlore) (figure 23.11
). Les OC sont connus pour exercer une action inhibitrice aiguë sur le système GABAergique, mais ils sont susceptibles de perturber le système dopaminergique (Rivera et coll., 1998
).
L’hydrophobicité importante des organochlorés leur permet de passer facilement la BHE. Ils s’accumulent dans certaines zones du cerveau. Des études rapportent une plus grande concentration de DDT (DichloroDiphénylTrichloroéthane) et de son principal métabolite, le DDE (DichloroDiphényldichloroEthylène) dans le cerveau post-mortem de patients atteints de maladie de Parkinson (Fleming et coll., 1994
; Corrigan et coll., 2000
), mais peu d’études expérimentales ont permis de démontrer un rôle des dichlorodiphényléthanes dans la pathologie. Ces remarques s’appliquent aussi aux hexachlorocyclohexanes (Corrigan et coll., 1998
). Des études sur la BHE montrent également qu’un OC (lindane) augmente la perméabilité de la barrière chez des rats exposés à des doses égales à 1/50
e de la LD50 (Gupta et coll., 1999
; Chan et coll., 2006a
et b
). Parmi les cyclodiènes, la dieldrine qui est aussi très hydrophobe, a été détectée dans les cerveaux post-mortem de personnes atteintes de la maladie de Parkinson (Fleming et coll., 1994
; Corrigan et coll., 1998
) notamment dans le noyau caudé, cible des neurones dopaminergiques de la substance noire.
Le transport de ces pesticides au niveau de la membrane plasmique a fait l’objet d’études plus mécanistiques portant sur l’importance des polymorphismes des transporteurs de xénobiotiques sur l’incidence de la maladie de Parkinson (Drozdzik et coll., 2003
; Lee et coll., 2004
; Bartels et coll., 2009
; Westerlund et coll., 2009
; Zschiedrich et coll., 2009
; Dutheil et coll., 2010
). Ces transporteurs sont des protéines de la membrane plasmique permettant un efflux actif (consommation d’ATP) d’un grand nombre de molécules (médicaments, métabolites, polluants…). L’un des transporteurs clés est la protéine P-gp (P-glycoprotéine), membre de la superfamille des ABC (
ATP binding cassette), codée par le gène
ABCB1 ou
MDR1 (
MultiDrug Resistance protein 1 gene). Elle est notamment exprimée au niveau de la barrière hémato-encéphalique (BHE, cellule endothéliale) et pourrait être importante pour permettre l’élimination de contaminants au niveau central. Bien que toutes les études ne montrent pas nécessairement une association, des publications récentes indiquent qu’un polymorphisme de la P-gp (G2677 associé à une diminution d’activité de la protéine, mesurée
in vitro) modifie de manière positive l’association entre exposition à des OCs et la maladie de Parkinson (Dutheil et coll., 2010
). En parallèle de ces problématiques de transport membranaire, le métabolisme des OCs module également les risques de développement de la maladie en cas d’exposition à ces pesticides. Elbaz et coll. (2004
) montrent ainsi qu’un faible métabolisme lié à l’activité du cytochrome p450 CYP2D6 est associé à un risque augmenté de développement d’une maladie de Parkinson (Elbaz et coll., 2004
).
Malgré cette prise en charge métabolique, les OCs comme la dieldrine sont très persistants dans l’environnement (25 ans pour la dieldrine) et les organismes (Homme : demi-vie comprise entre 250-400 jours pour la dieldrine) du fait de leur grande hydrophobicité. Les concentrations sanguines de dieldrine peuvent atteindre 5-15 µg/l. Elle peut aussi être produite métaboliquement à partir de l’aldrine, un autre cyclodiène. La dieldrine peut ensuite être métabolisée par hydrolyse ou oxydation enzymatique.
Mécanismes d’action
Une intoxication aiguë par de la dieldrine provoque nausées, maux de tête, vomissement, convulsions et coma (en fonction de la dose). La dose létale chez l’Homme est comprise entre 1,5 et 5 g. Le système nerveux central est une cible majeure de la dieldrine qui semble inhiber la fonction de certains canaux ou récepteurs membranaires dont ceux du GABA (
Gamma AminoButyric Acid) (Vale et coll., 2003
; Zhao et coll., 2003
; Kanthasamy et coll., 2005
). L’inhibition du récepteur GABA-A provoque une hyperexcitabilité neuronale et notamment une entrée massive de Ca
2+ au niveau des récepteurs glutamatergiques (Pomes et coll., 1994
; Ikeda et coll., 1998
; Vale et coll., 2003
) (figure 23.12
).
De nombreuses études ont été conduites pour déterminer si l’exposition chronique aux OC pouvait favoriser le développement d’une maladie de Parkinson comme le suggèrent les études analytiques. La dieldrine est sélectivement neurotoxique vis-à-vis des neurones dopaminergiques de la substance noire comparativement aux cellules GABAergiques de même localisation (Sanchez-Ramos et coll., 1998
; Kitazawa et coll., 2001
) et elle potentialise les effets neurotoxiques du MPTP (Richardson et coll., 2006
). Une exposition alimentaire à de basses concentrations de dieldrine provoque une diminution significative de 60 % des niveaux de dopamine dans le cerveau de colombes probablement par neurodégénérescence des neurones dopaminergiques (Heinz et coll., 1980
). Cet effet a été reproduit sur des canards et des rats (Sharma et coll., 1976
; Wagner et Greene, 1978
). Cette diminution des concentrations intracellulaires de dopamine a été observée sur cellules dopaminergiques en culture (PC12) (Kitazawa et coll., 2001
). À l’origine de la neurodégénérescence, l’un des mécanismes activés par les OC pourrait être une augmentation des concentrations cytoplasmiques de dopamine, qui a une action intrinsèque pro-oxydante. L’exposition de souris à de l’heptachlore augmente ainsi l’expression du transporteur de la dopamine (DAT qui favorise la ré-absorption du neurotransmetteur) et du transporteur vésiculaire des monoamines (VMAT2 qui permet l’accumulation dans les vésicules de sécrétion par transfert en provenance du cytoplasme) dans le striatum, tout en inhibant la fonction de ce dernier (avec pour conséquence une accumulation de dopamine cytoplasmique) (Baker et coll., 1991
; Miller et coll., 1999
; Purkerson-Parker et coll., 2001
; Bloomquist et coll., 2002
; Kirby et coll., 2002
). Cette dérégulation pourrait provoquer une recapture cytoplasmique plus importante de dopamine par les terminaisons synaptiques et abolir le transfert vers les vésicules intracellulaires avec pour conséquence des niveaux de dopamine plus importants dans le cytoplasme. En l’absence de localisation vésiculaire, l’effet pro-oxydant de la dopamine pourrait alors endommager ces terminaisons. Tout comme l’heptachlore, la dieldrine est capable de perturber l’homéostasie de la dopamine en agissant sur le DAT (Richardson et coll., 2006
; Hatcher et coll., 2007
).
En plus d’une action sur la recapture de dopamine, les OC ont un effet sur le stress oxydant et la formation d’agrégats. Ils augmentent le stress oxydant dans des lignées cellulaires dopaminergiques (Chun et coll., 2001
; Kitazawa et coll., 2001
). D’un point de vue mécanistique, les OC inhibent le complexe 3 de la chaîne respiratoire et diminuent la production d’ATP (Kitazawa et coll., 2001
et 2003
; Kanthasamy et coll., 2005
; Hatcher et coll., 2007
) ce qui avait été évoqué dès 1971 (Bergen, 1971
). En plus de cet effet sur la mitochondrie et la production de DRO, la dieldrine stimule la formation de fibrilles d’alpha-synucléine et leur agrégation
in vitro (Kitazawa et coll., 2001
; Stedeford et coll., 2001
; Uversky et coll., 2001
et 2002
; Kanthasamy et coll., 2005
) ainsi que l’accumulation de protéines ubiquitinées par inhibition à long terme des voies de dégradation, dépendantes du protéasome.
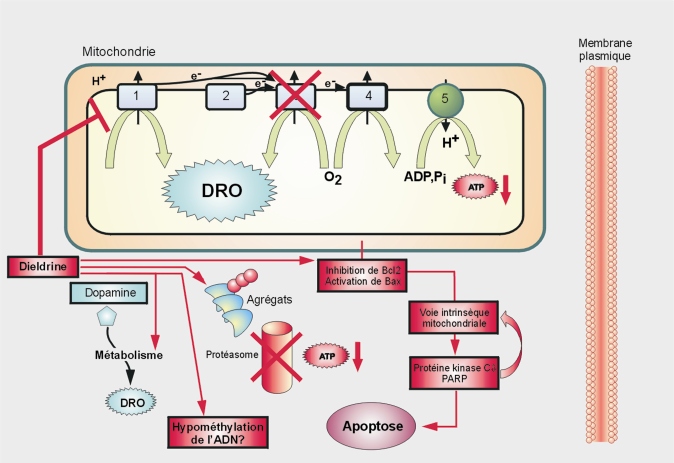
Les OC activent également l’apoptose des neurones dopaminergiques (Kanthasamy et coll., 2005
). L’étude de Kitazawa de 2001 (Kitazawa et coll., 2001
) montre que l’apoptose est associée à une libération de cytochrome C par la mitochondrie (PC12). Ce phénomène est inhibé par la surexpression de la protéine anti-apoptotique Bcl-2. Plusieurs études démontrent également une activation de la caspase 3 (Chun et coll., 2001
; Kitazawa et coll., 2003
). Deux cibles de la caspase 3 (la poly-ADP ribose polymérase ou PARP et la protéine kinase C∂
11
Les autres isoformes ne sont pas touchées.
) sont ainsi clivées en réponse à la dieldrine (Kitazawa et coll., 2003
et 2004
). L’apoptose des cellules traitées est bloquée à la fois par des anti-oxydants, des inhibiteurs de la PKC∂ et par un inhibiteur de la MAO, suggérant que la dopamine et son métabolisme sont impliqués dans la mort cellulaire (Kitazawa et coll., 2001
et 2003
). Le rôle déterminant de la PKC∂ est retrouvé dans l’apoptose induite par d’autres pesticides ou toxines (MMT, manganèse, MPP+). L’un des effets principaux de cette kinase serait d’amplifier la voie des caspases notamment la caspase 3 par un mécanisme encore non identifié (Anantharam et coll., 2002
; Kanthasamy et coll., 2005
). D’autres kinases ont depuis été identifiées dans les voies de signalisation activées par les OC comme les JNK, les MAPK ou la p110 kinase (Chun et coll., 2001
; Shinomiya et Shinomiya, 2003
). L’activation des MAPK dans l’étude de Shinomiya suggère une altération des processus de différenciation. Cette étude suggère toutefois que le DDT a un effet pro-apoptotique plus marqué que son métabolite le DDE, ce qui laisse sous-entendre que l’activité des enzymes métabolisant les pesticides (composante génétique individuelle) est importante à considérer (Shinomiya et Shinomiya, 2003
).
Concernant les effets à long terme sur l’expression génique, une étude récente menée sur 86 individus sains, a montré que les taux de cinq OC (heptachlore epoxide, oxychlordane, trans-nonachlore, DDE et DDT) dans le sérum étaient inversement proportionnels à une hypométhylation générale de l’ADN (mesurée en utilisant les éléments Alu et Line-1) (Kim et coll., 2010
). La méthylation de l’ADN régule l’expression des gènes et est associée à différents processus dont la cancérisation. L’intérêt de cette étude porte à la fois sur des échantillons humains, des doses de pesticides pertinentes, et l’identification d’une modification épigénétique qui pourrait affecter les neurones et conduire à des perturbations de l’expression de gènes.
En résumé, les organochlorés hydrophobes passent facilement la barrière BHE. La dieldrine est sélectivement neurotoxique vis-à-vis des neurones dopaminergiques de la substance noire. Le mécanisme d’action pourrait être une augmentation des niveaux de dopamine dans le cytoplasme et un effet pro-oxydant endommageant les terminaisons. L’heptachlore et la dieldrine perturbent l’homéostasie de la dopamine. Par ailleurs, une modification épigénétique (hypométhylation) pourrait affecter les neurones et conduire à des perturbations de l’expression de gènes.
Organophosphorés (chlorpyrifos, diazinon, malathion, parathion)
Les organophosphorés (OPs) sont des insecticides qui ciblent l’acétylcholine estérase (AChE), enzyme exprimée dans le système nerveux central et périphérique, qui hydrolyse et inactive l’acétylcholine (figure 23.13
). L’acétylcholine (ACh) est un neurotransmetteur important aussi bien dans le système nerveux central, où elle est impliquée dans la mémoire et l’apprentissage, que dans le système nerveux périphérique, notamment dans l’activité musculaire et les fonctions végétatives. L’acétylcholine se fixe sur deux types de récepteurs : les récepteurs nicotiniques (canaux cationiques provoquant en quelques millisecondes après activation par l’acétylcholine, une dépolarisation et une excitation) et les récepteurs muscariniques, couplés aux protéines G, plus lents, peuvent induire des réponses excitatrices ou inhibitrices. L’acétylcholine joue un rôle fondamental dans le développement du cerveau (Slotkin, 2004
). L’acétylcholine est ainsi susceptible de favoriser la réplication des précurseurs neuronaux, leur différenciation, la régulation de la synaptogenèse, de l’apoptose et des processus migratoires. Plusieurs polluants (par exemple : nicotine du tabac en exposition passive ou active) ont été caractérisés comme des perturbateurs du système cholinergique contribuant ainsi à des perturbations neuro-développementales. Par ailleurs, plusieurs études épidémiologiques (notamment celles menées chez des éleveurs de mouton utilisant les OPs pour éliminer les parasites
12
Et utilisés en remplacement des organochlorés
) suggèrent que l’exposition chronique à des faibles concentrations d’OPs conduit à l’apparition de déficits neurocomportementaux classés en trois catégories
13
Inhibition aiguë de l’AchE ; effets retardés de l’inhibition de l’AChE ou syndrome intermédiaire ; polyneuropathie retardée.
.
Mécanismes d’action
Peu d’études ont analysé l’influence des organophosphorés sur le développement d’une maladie de Parkinson. Ils seraient susceptibles de diminuer la recapture de la dopamine au niveau striatal (Karen et coll., 2001
) ou de perturber la structure du cytosquelette par divers mécanismes (hyperphosphorylation, activation de la calpaïne). Par ailleurs, des effets communs à ceux d’autres pesticides ont été décrits comme le ciblage de certains complexes de la chaîne respiratoire (I et IV) de la mitochondrie. Par extrapolation, on peut poser l’hypothèse d’un effet sur la production d’ATP (Hargreaves, 2012
).
En résumé, bien que certains mécanismes puissent évoquer un impact des OP sur le système dopaminergique, leur implication dans le développement d’une maladie de Parkinson reste à démontrer.
Pyréthrinoïdes (perméthrine)
Les pyréthrinoïdes sont des insecticides dérivés des pyréthrines de chrysanthème (figure 23.14
).
Les intoxications aiguës sont rares chez l’Homme et provoquent vertiges, fatigues et maux de tête (coma dans les cas les plus sévères) (Bradberry et coll., 2005
).
Mécanismes d’action
La toxicité aiguë est principalement due au maintien de l’activation de canaux sodium voltage-dépendants, qui perturberait un très grand nombre de mécanismes de signalisation. Ainsi, les pyréthrinoïdes inhibent la signalisation GABAergique, modulent la transmission cholinergique, favorisent la libération de noradrénaline et agissent sur les canaux calciques et chlore. Tous ces effets pourraient être liés à leur liaison directe sur les canaux sodium voltage-dépendants (Ray et Fry, 2006
). Deux classes de pyréthrinoïdes existent et ne présentent pas les mêmes propriétés vis-à-vis des canaux précités (Bjorling-Poulsen et coll., 2008
).
Quelques études expérimentales suggèrent un rôle de la perméthrine dans le développement d’une maladie de Parkinson. Une administration régulière de perméthrine à des souris pendant 2 semaines (3 fois, 0,8 mg/kg) augmente la recapture de la dopamine de 30 % après le dernier traitement (Gillette et Bloomquist, 2003
; Elwan et coll., 2006
). Cet effet pourrait dépendre de la dose utilisée (diminution de la recapture à forte dose) (Karen et coll., 2001
). L’implication du transporteur de la dopamine (DAT) n’a pourtant pas été clairement démontrée par les auteurs (et par d’autres études indépendantes) (Karen et coll., 2001
; Bloomquist et coll., 2002
; Pittman et coll., 2003
; Nasuti et coll., 2007
) comme évoqué pour l’action de l’heptachlore qui augmente son expression dans le striatum de souris C57BL exposées (Bloomquist et coll., 2002
). De hautes concentrations de perméthrine favorisent la surexpression de GFAP (
Glial Fibrillary Acidic Protein) par les cellules gliales du striatum, témoin d’une neuro-inflammation (Pittman et coll., 2003
). Des cultures cellulaires de neuroblastome (SH-SY5Y) exposées à diverses doses de pyréthrinoïdes (perméthrine, cyperméthrine, pyréthrine) ont permis de déterminer par la mesure des concentrations en ATP, les seuils de toxicité de ces molécules (cyperméthrine > perméthrine > pyréthrine) (Kakko et coll., 2004
). Des expériences menées sur d’autres régions du cerveau (cortex) chez des rats exposés à la deltaméthrine ont montré des expressions anormalement prolongées des facteurs de transcription c-Fos et c-Jun (associées préalablement à des processus neurodégénératifs) dépendantes de l’activité de récepteurs glutamatergiques (AMPA) (Wu et Liu 2003
). La substance noire n’a pas été examinée dans cette étude.
En résumé, quelques travaux expérimentaux semblent en faveur du rôle de la perméthrine dans le développement d’une maladie de Parkinson en montrant une augmentation de la recapture de la dopamine à faibles doses.
Chlorophénoxyherbicides (2,4-D)
Les chlorophénoxyherbicides sont utilisés pour contrôler les mauvaises herbes dans les cultures de céréales. En cas d’intoxication aiguë, les effets sont variables allant des convulsions au coma (Bradberry et coll., 2005
).
Nature chimique de la molécule, métabolisme, passage de la barrière hémato-encéphalique
Les chlorophénoxyherbicides comportent une partie carbonée aliphatique acide (COOH) et une partie aromatique substituée par des atomes de chlore (figure 23.15
).
Les chlorophénoxyherbicides sont hydrolysés ou dissociés
in vivo. La toxicité des composés semble dépendre principalement de la partie aliphatique acide. Ils lient l’albumine notamment ceux à chaînes longues et comportant une partie aromatique très substituée. Leur biodisponibilité est donc très variable en fonction de leur nature chimique. De faibles concentrations de chlorophénoxyherbicides sont retrouvées dans le cerveau de modèles animaux exposés à des doses inférieures à 100 mg/kg (Elo et Ylitalo, 1979
). À ces doses, ils perturbent peu les membranes dont la BHE. À des doses supérieures (>250 mg/kg), la BHE subit des dommages réversibles et les chlorophénoxyherbicides sont retrouvés dans le cerveau (Hervonen et coll., 1982
).
Mécanismes d’action
Les mécanismes d’action des chlorophénoxyherbicides demeurent inconnus, ils seraient responsables d’une perturbation de l’intégrité des membranes cellulaires. Le 2,4-D est le chlorophénoxyherbicide le plus utilisé.
In vitro, il perturbe fortement les membranes à des concentrations supérieures à 10 µM. Les chlorophénoxyherbicides perturbent les systèmes de transport membranaires notamment ceux du plexus choroïde responsables de la détoxication partielle de certains anions toxiques (métabolites de certains neurotransmetteurs). Des dérivés de la dopamine peuvent ainsi s’accumuler dans le SNC à la suite d’un traitement par le 2,4-D (Elo et MacDonald, 1989
). Ceci va de pair avec les effets d’une injection de 2,4-D dans le striatum de rats qui provoque une diminution importante de l’activité locomotrice et des modifications des concentrations de plusieurs monoamines dans différentes parties du cerveau de rats. Les niveaux de dopamine mais aussi de certains récepteurs (D2) sont ainsi affectés avec des résultats variables en fonction du protocole d’exposition, de la durée et de la continuité du traitement (Bortolozzi et coll., 1999
, 2001
et 2004
). Ces effets pourraient être réversibles. Une co-exposition, 2,4-D et acide 2,4,5-trichlorophénoxyacétique, chez des rattes gestantes (entre 6 et 15 jours de gestation) entraîne des anomalies de développement du système dopaminergique et du comportement associé (Mohammad et St Omer, 1985
). Il serait intéressant d’examiner les membranes mitochondriales de cellules dopaminergiques exposées au 2,4-D.
Le 2,4-D pourrait être à l’origine d’un stress oxydant (expérience menée
in vitro sur des cellules granulaires de cervelet) comme l’attestent les baisses d’activité catalase et la diminution des concentrations de glutathion (Bongiovanni et coll., 2007
).
Ces différents mécanismes (perturbation membranaire, stress oxydant) pourraient être à l’origine d’une apoptose des cellules dopaminergiques.
En résumé, le mécanisme d’action des chlorophénoxyherbicides demeure inexpliqué. Des anomalies de développement du système dopaminergique et du comportement associé ont été signalées après exposition chez des femelles (rat) gestantes. Les modifications au niveau des membranes mitochondriales de cellules dopaminergiques exposées au 2,4-D mériteraient d’être explorées.
Autres pathologies neurodégénératives et troubles du neurodéveloppement : lien avec les pesticides
Organochlorés
Tout comme pour la maladie de Parkinson, une augmentation de marqueurs du stress oxydant est observée dans les cerveaux post-mortem de patients atteints de maladie d’Alzheimer. Dans la lignée de ces observations, des concentrations plus élevées de pp-DDT sont retrouvées dans les cerveaux post-mortem de patients (par opposition à des témoins) (Fleming et coll., 1994
).
Organophosphorés (OPs)
Comme indiqués préalablement (paragraphe Parkinson), les OPs ciblent l’acétylcholine estérase (AChE), enzyme exprimée dans le système nerveux central et périphérique, qui hydrolyse et inactive l’acétylcholine (celle-ci joue un rôle dans la mémoire, l’apprentissage, l’activité musculaire, le développement du cerveau et au niveau cellulaire, la réplication des précurseurs neuronaux, leur différenciation, la régulation de la synaptogenèse, de l’apoptose et des processus migratoires). L’exposition chronique à des faibles concentrations d’OPs conduit à l’apparition de trois types d’effets : inhibition aiguë de l’AChE (intoxication aiguë) ; effets retardés de l’inhibition de l’AChE ou syndrome intermédiaire ; poly-neuropathie retardée.
Dans le syndrome cholinergique, les OPs forment une liaison covalente avec le site actif de l’AChE (avec une sérine) et sont donc des inhibiteurs irréversibles de l’enzyme (Lauder et Schambra, 1999
). La conséquence directe d’une exposition aux OPs est l’augmentation des concentrations d’acétylcholine dans la fente synaptique et la stimulation trop importante des récepteurs muscariniques et nicotiniques. Une intoxication aiguë aux OPs conduit à un syndrome cholinergique avec, en fonction du degré d’empoisonnement, maux de tête, vomissements, confusion, ataxie, coma et blocage respiratoire. Ces manifestations cliniques n’apparaissent qu’au-delà d’un seuil d’inhibition de 70 %, laissant supposer des effets chroniques sous le seuil de détection.
Des effets retardés (OPIDN ou
OP-Induced Delayed Neuropathies, voir Hargreaves, 2012
) sont observés par atteinte d’une estérase particulière, la NTE (
Neuropathy Target Esterase), enzyme principalement exprimée dans le système nerveux (Lotti, 1991
; Johnson et Glynn, 1995
; Lotti et Moretto, 2005
). La période asymptomatique qui suit l’intoxication, dure quelques jours à quelques semaines. Les symptômes peuvent être réversibles mais des séquelles potentielles (déficits moteurs) peuvent persister comme le suggèrent des études menées chez le rat (Pope et coll., 1992
; Sanchez-Santed et coll., 2004
). La NTE est impliquée dans le métabolisme phospholipidique (type phospholipase B). L’invalidation complète de NTE est létale. En revanche, les animaux présentant une invalidation restreinte à certaines zones du cerveau survivent et présentent des signes de neurodégénérescence (des résultats similaires se retrouvent chez la drosophile). Au niveau cellulaire, l’enzyme est ancrée au réticulum endoplasmique où elle forme un canal ionique dont la dérégulation de la fonction semble essentielle pour l’apparition des neuropathies. Elle pourrait également jouer un rôle dans l’hydrolyse de certains phospholipides dont la phosphatidylcholine (Hargreaves, 2012
). Les études d’invalidation sur modèles cellulaires sont quelque peu discordantes mais suggèrent un rôle potentiel de la NTE dans la différenciation neuronale. Hormis la NTE, d’autres marqueurs ont été identifiés dans les effets retardés de certains OPs : une hyperphosphorylation de certaines protéines du cytosquelette (par activation potentielle de la calmoduline kinase ou de ERK1/2) ce qui pourrait perturber la formation des neurites, l’activation de la calpaïne, une protéase dégradant certaines protéines des neurofilaments, et/ou une altération de l’activité de certaines protéines mitochondriales de la chaîne respiratoire (complexes I et IV).
Les OPs inhibent également à des degrés variables, la butyrylcholinestérase (ou cholinestérase plasmatique dont la mesure d’activité fournit facilement des données sur l’exposition, tout comme celle de l’AChE érythrocytaire).
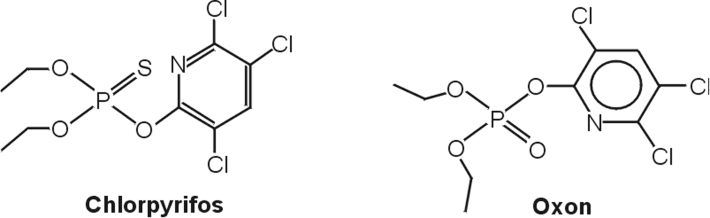
Les OPs nécessitent pour la plupart une activation par remplacement de la fonction P=S par une fonction P=O (par exemple : le chlorpyrifos est métabolisé par le foie en chlorpyrifos oxon). Il existe une variabilité du métabolisme des OPs liée aux polymorphismes des enzymes du métabolisme des xénobiotiques notamment ceux de la paraoxonase 1 (PON1), une enzyme qui hydrolyse les métabolites actifs des OPs (Costa et coll., 2003
). Le chlorpyrifos, tout comme l’ensemble des phosphorothioates (par exemple : diazinon), a ainsi peu d’activité en tant que tel, vis-à-vis de l’AChE. Il est soit activé en chlorpyrifos oxon principalement par les cytochromes P450 (CYP), le CYP2B6 (et dans une moindre mesure le CYP1A2) ; soit inactivé principalement par les CYP2C19 et 2C9 (Sams et coll., 2004
).
Ces CYP font l’objet de polymorphismes et une forte activité des CYP2B6 et 1A2 associée à une faible activité de CYP2C19, pourrait donc résulter en une forte toxicité du chlorpyrifos. Si l’on considère l’ensemble des OPs, l’activité des CYP peut varier d’un composé à l’autre. Ainsi, le diazinon est métabolisé en diazoxon de manière équivalente par les CYP2B6 et 1A2 (Povey, 2010
). Par ailleurs, le CYP3A4 bien que contribuant peu par unité enzymatique à l’activation du chlorpyrifos, est toutefois fortement exprimé dans le foie et pourrait à ce titre, être une enzyme majeure du métabolisme des OPs du fait de son niveau d’expression.
La paraoxonase 1 (PON1) est une enzyme plasmatique et hépatique, qui inactive les métabolites oxon (Furlong, 2007
). Les animaux invalidés pour cette enzyme présentent une plus grande susceptibilité vis-à-vis des OPs. Il existe une forte variabilité interindividuelle chez l’Homme de l’activité plasmatique de PON1 (Geldmacher, 1998
; Costa et coll., 2003
) qui serait liée à des polymorphismes localisés dans la partie codante et non codante du gène. Parmi les polymorphismes plus importants, on recense :
• (C-108T) dans le promoteur qui détermine le niveau d’expression (régulation transcriptionnelle) (Brophy et coll., 2001
) ;
• (Q192R) dans la partie codante.
Ces polymorphismes doivent être considérés avec précision pour l’ensemble des OPs. Ainsi, Davies et coll. rapportent que l’isoforme 192R a une activité plus basse vis-à-vis du diazoxon (métabolite du diazinon) que celle vis-à-vis du chlorpyrifos (Davies et coll., 1996
). Ce résultat est toutefois controversé (O’Leary et coll., 2005
; Mutch et coll., 2007
; Richter et coll., 2009
). L’utilisation de modèles animaux pour différencier l’importance des polymorphismes pourrait être délicate. Les souris KO pour PON1 perfusées avec soit PON1-192R, soit PON1-192Q présentent les mêmes métabolismes vis-à-vis du diazoxon. En revanche, l’isoforme R est plus protectrice vis-à-vis du chlorpyrifos (Li et coll., 2000
). Ces résultats ont été confirmés depuis par expression transgénique de la PON1 humaine 192Q ou 192R sur souris humanisées (Cole et coll., 2005
).
Le rôle de ces polymorphismes a été décortiqué chez la souris par l’utilisation d’animaux KO ou transgéniques vis-à-vis des deux isoformes humaines (sur un fond génétique PON1 KO). L’analyse transcriptomique (
Gene Set Analysis) des cervelets de ces animaux (exposés entre les jours 4 et 21 après la naissance à des doses de chlorpyrifos comprises entre 0,35 et 0,50 mg/kg/jour) révèle une modification des fonctions cellulaires suivantes : fonctionnement mitochondrial, stress oxydant, neurotransmission, développement du système nerveux central. Différentes signatures géniques sont identifiées chez les souris R192 ou Q192. Enfin, l’activité de l’AChE est diminuée par ces expositions répétées chez la souris KO ou Q192 mais pas chez la souris sauvage ou R192 (qui hydrolyse mieux le chlorpyrifos oxon). L’ensemble de ces données suggère que des expositions chroniques aux OPs (ici le chlorpyrifos) n’affectent pas de manière similaire chaque individu (en fonction de son génotype) et sont susceptibles d’interférer avec des processus cellulaires essentiels à l’homéostasie neuronale (Cole et coll., 2011
).
La régulation transcriptionnelle du gène PON1 est également dépendante de facteurs environnementaux dont les pesticides (Costa et coll., 2003
; Hernandez et coll., 2003
) et potentiellement les polyphénols de l’alimentation (Gouedard et coll., 2004
). La détoxication des OPs dépend d’autres protéines dont les carboxyestérases, l’AChE, la BChE et les glutathion S-transférases (GST) (Povey et coll., 2007
).
Concernant les anomalies du développement associées à l’exposition aux OPs, le chlorpyrifos est le plus utilisé pour caractériser les effets mécanistiques de cette famille de molécules. Avant son interdiction dans les produits domestiques et de jardinages aux États-Unis, des cas d’exposition supérieure à la normale ont été rapportés chez des femmes enceintes et des enfants. L’utilisation de modèles murins a permis de montrer que l’exposition prénatale ou néonatale au chlorpyrifos s’accompagne d’altérations des performances locomotrices ou cognitives (hyperactivité motrice, troubles des apprentissages et de la mémoire) (Ricceri et coll., 2003
; Icenogle et coll., 2004
; Ricceri et coll., 2006
). À des concentrations comparables à celles mesurées dans le méconium (premières selles de l’enfant), le chlorpyrifos induit des anomalies mitotiques, des signaux apoptotiques des cellules en culture du tube neural d’embryons de rat (Roy et coll., 1998
).
Des marqueurs de stress oxydants sont également observés chez des travailleurs exposés (Ranjbar et coll., 2002). Ces données s’ajoutent à celles montrant qu’une exposition
in utero (rats) provoque des déficits du nombre de cellules cérébrales, des projections neurales et de la communication synaptique (Qiao et coll., 2002
, 2003a
et b
et 2004
). La période de sensibilité concerne également le développement post-embryonnaire : la gliogenèse et la synaptogenèse ont lieu majoritairement dans les 2-3 premières semaines qui suivent la naissance chez le rat (équivalente à celle du développement néonatal chez l’Homme) (Moser, 2000
; Levin et coll., 2001
; Vidair, 2004
). Pendant la période de différenciation sexuelle du cerveau, le chlorpyrifos perturbe l’expression des récepteurs sérotoninergiques et la connexion des neurones correspondants avec leurs cibles ce qui est à l’origine de symptômes évoquant un déficit en sérotonine.
Sur les cibles cellulaires du chlorpyrifos et de son métabolite, les anomalies provoquées par celui-ci au cours de la période prénatale sont parfois observées à des doses inférieures à celles nécessaires pour inactiver l’AChE (Qiao et coll., 2003
; Ricceri et coll., 2003
et 2006
; Icenogle et coll., 2004
; Moreira et coll., 2010
) suggérant un mécanisme d’action indépendant de l’AChE, passant par la protéine kinase A (Huff et coll., 1994
; Ward et Mundy 1996
; Song et coll., 1997
; Garcia et coll., 2001
; Olivier et coll., 2001
; Schuh et coll., 2002
; Zhang et coll., 2002
), la protéine kinase C (Bomser et Casida, 2000
; Bomser et coll., 2002
), les facteurs de transcription c-fos, p53, AP-1, Sp1 et CREB (Crumpton et coll., 2000
; Garcia et coll., 2001
; Schuh et coll., 2002
; Dam et coll., 2003
; Slotkin, 2004
) ou certaines protéines du cytosquelette (tubuline). Une étude récente montre que sur des coupes de striatum en culture, un traitement par le chlorpyrifos (100 µM) stimule la phosphorylation de cibles de la PKA (DARPP-32 ou la sous-unité GluR1 du récepteur AMPA du glutamate) et de la protéine tau. Le chlorpyrifos semble favoriser la libération de glutamate au niveau striatal ce qui pourrait contribuer au phénomène d’excitotoxicité (Torres-Altoro et coll., 2011
). Les effets indépendants de l’AChE et dépendants de la voie PKA ont été retrouvés en utilisant des cultures de cellule (PC12). De manière intéressante, l’exposition de cellules différenciées réduit les activités adénylate cyclase (enzyme produisant l’AMPc, activateur de la PKA) après 2 jours de traitement mais augmente ces activités au-delà de 6 jours de traitement (en continu ou après 2 jours de traitement avec chlorpyrifos puis 4 jours sans traitement) (Adigun et coll., 2010a
et b
). Par ailleurs, certains effets du chlorpyrifos sont observés à des stades embryonnaires pour lesquels l’AChE n’est pas encore exprimée (Lauder et Schambra, 1999
), dans des zones du cerveau exprimant très peu d’AChE (Whitney et coll., 1995
) ou en utilisant des injections directes du chlorpyrifos sans métabolisation hépatique ce qui renforce l’hypothèse d’un mécanisme indépendant n’impliquant pas le métabolite « oxon » (Campbell et coll., 1997
; Dam et coll., 1998
, 2000
et 2003
; Johnson et coll., 1998
; Levin et coll., 2001
; Slotkin et coll., 2001
et 2002
). Une étude transcriptomique (
Affymetrix Mouse Whole Genome) récente réalisée sur des souris gestantes et exposées à différentes doses de chlorpyrifos pendant la période prénatale (2-15 mg/kg/jour, jours 6-17) révèle par ailleurs que les cibles géniques de ce pesticide sont parfois communes et parfois différentes chez la mère et chez le fœtus (détails de l’analyse GO Stat dans Moreira et coll., 2010
). Les modifications d’expression génique sont d’ailleurs obtenues à des doses inférieures à la LOAEL dans cette étude suggérant un mécanisme d’action différent de celui impliquant un ciblage de l’AChE.
La glie pourrait être une cible du chlorpyrifos comme le montrent plusieurs études (Whitney et coll., 1995
; Campbell et coll., 1997
; Dam et coll., 1998
; Monnet-Tschudi et coll., 2000
; Garcia et coll., 2001
et 2002
; Qiao et coll., 2001
). Cette diversité des cibles des OPs suggère que deux composés d’une même famille pourraient activer des gènes communs mais aussi différentes catégories géniques conduisant ainsi à des manifestations pathologiques sensiblement différentes. Une étude transcriptomique utilisant le chlorpyrifos et le diazinon confirme cette hypothèse (Slotkin et coll., 2007
, 2008a
et b
).
La neurotoxicité des OPs et de leurs métabolites pourrait être liée à l’activation de l’apoptose comme le montre une étude menée avec des neurones primaires de rat en culture. L’effet du chlorpyrifos, dans cet article, serait toujours indépendant de l’inhibition de l’AChE, et passerait par une activation des kinases ERK1/2 et p38. Toutefois, l’utilisation d’inhibiteurs pharmacologiques de cette voie suggère un tableau plus compliqué que ce que laisse sous-entendre leur activation commune par le chlorpyrifos et son métabolite le chlorpyrifos oxon. En effet, ERK1/2 semble exercer une action pro-apoptotique alors qu’à l’inverse, p38 semble protéger la cellule de la dégénérescence. Par ailleurs, seule une fraction des JNK (dans le noyau) est activée par le chlorpyrifos et contribue au phénomène d’apoptose par phosphorylation de c-Jun, un facteur de transcription (Caughlan et coll., 2004
).
En résumé, l’implication des OP dans des dégénérescences neuronales et anomalies du développement semble plausible. L’exposition prénatale ou néonatale au chlorpyrifos conduit à l’altération des performances locomotrices ou cognitives dans des modèles murins. Le chlorpyrifos induit des anomalies mitotiques, des signaux apoptotiques des cellules en culture du tube neural d’embryons de rat. Les OP pourraient agir par différents mécanismes certains n’impliquant pas l’AChE. En effet, certains effets du chlorpyrifos sont observés à des stades embryonnaires pour lesquels l’AChE n’est pas encore exprimée. L’effet des métabolites « oxon » est également reconnu et le rôle des polymorphismes PON1 a été confirmé dans des modèles transgéniques.
Carbamates et dithiocarbamates
Les carbamates ciblent l’AChE mais leur inhibition est réversible
14
Le paraquat pourrait aussi agir sur cette enzyme (Alcaro et coll., 2007).
. Ils conduisent à une stimulation cholinergique exacerbée mais qui ne dure que quelques minutes à quelques heures. Les symptômes d’intoxication aiguë diffèrent peu de ceux provoqués par les OPs chez les enfants. En revanche, l’hypotonie et les diarrhées ne sont pas retrouvées chez les adultes qui présentent d’autres signes cliniques comme une myosis (rétrécissement de la pupille) et des fasciculations (contractions spontanées, involontaires et irrégulières de faisceaux musculaires).
L’un des modes d’action couramment décrits pour les carbamates est une augmentation du stress oxydant (Gupta et coll., 2007
). Le carbofuran provoque ainsi une augmentation de la peroxydation lipidique et une diminution des défenses anti-oxydantes dans des cerveaux de rats exposés. La physostigmine (alcaloïde inhibant l’AChE) inhibe la synthèse de l’ADN et augmente la peroxydation lipidique dans des cellules PC12 en différenciation (Slotkin et coll., 2007
).
Les dithiocarbamates (EBDC) semblent perturber le transport vésiculaire du glutamate, l’un des neurotransmetteurs les plus importants dans le SNC (Vaccari et coll., 1999
). L’exposition à certains EBDCs provoque par ailleurs des malformations fœtales chez l’animal rappelant une inhibition de la fonction thyroïdienne. Un faible hypothyroïdisme peut conduire à des déficits intellectuels détectables chez les enfants. L’inhibition de la synthèse des hormones thyroïdes par l’ETU qui est un métabolite pourrait donc être un sujet de préoccupation (Doerge et Takazawa, 1990
; Marinovich et coll., 1997
). L’effet combiné du manèbe et du paraquat pourrait avoir des effets additifs sur cette fonction.
En résumé, l’impact des carbamates et dithiocarbamates sur le SNC paraît plausible par une augmentation du stress oxydant pour les premiers ou perturbation du transport vésiculaire du glutamate pour les seconds.
Chlorophénoxyherbicides
Des défauts de myélinisation ou du développement du SNC accompagnés parfois d’anomalies comportementales ont été observés chez des poulets ou des rats en cas d’exposition pendant la période de développement (Duffard et coll., 1996
; Bortolozzi et coll., 1999
et 2004
; Ferri et coll., 2007
).
Comme évoqué préalablement, le 2,4-D peut provoquer des perturbations au niveau des systèmes de transport membranaires et conduire à l’accumulation d’anions toxiques dans le SNC. En plus de la dopamine, la sérotonine et les acides homovanilliques et 5-hydroxy-3indolacétiques peuvent ainsi s’accumuler (Elo et MacDonald, 1989
). D’autres perturbations au niveau de l’homéostasie des neurotransmetteurs peuvent survenir en cas d’exposition au 2,4-D. Celui-ci par analogie structurale avec l’acétyl-coA (précurseur ou impliqué dans la biogenèse de l’acétylcholine) pourrait ainsi inhiber la synthèse endogène d’acétylcholine (Bradberry et coll., 2000
).
Le 2,4-D perturbe également la neuritogenèse
in vitro par blocage de la polymérisation des microtubules (Rosso et coll., 2000
) ou comme évoqué précédemment, entraîne une apoptose des cellules granulaires de cervelet et une inhibition de la fonction thyroïdienne.
En résumé, les chlorophénoxyherbicides (2,4-D) sont susceptibles d’agir au niveau des systèmes de transport membranaires, de l’homéostasie des neurotransmetteurs, de la neuritogenèse. Ils entraînent une apoptose des cellules granulaires de cervelet et une inhibition de la fonction thyroïdienne.
Pyréthrinoïdes
Les jeunes rats sont 4 à 17 fois plus sensibles que les rats adultes à l’exposition des pyréthrinoïdes à hautes doses, probablement en raison d’un métabolisme de détoxication moins important (Cantalamessa, 1993
; Sheets et coll., 1994
; Sheets, 2000
). Cette différence de sensibilité pourrait être également liée à un effet des pyréthrinoïdes sur des canaux sodium voltage-dépendant dont les isoformes sont différents chez le jeune et l’adulte (Shafer et coll., 2005
). Ces canaux présentent des mutations (responsables d’épilepsie) qui pourraient expliquer des différences interindividuelles (Meisler et coll., 2001
). Les effets des pyréthrinoïdes se rapprochent de ceux de la phénytoïne, un anti-convulsant qui bloque les canaux sodiques (Adams et coll., 1990
). Des souris exposées en période postnatale (J10 à 16) à la phénytoïne ont une activité motrice plus importante et un défaut d’apprentissage. La densité des récepteurs muscariniques est modifiée chez ces animaux (Eriksson et Nordberg 1990
; Eriksson et Fredriksson 1991
; Malaviya et coll., 1993
; Ahlbom et coll., 1994
; Talts et coll., 1998a
et b
; Aziz et coll., 2001
). Dans ce contexte, d’autres études ont montré des changements de comportements sexuels (Lazarini et coll., 2001
), d’apprentissage (Moniz et coll., 1990
), d’activité motrice et de perméabilité de la BHE. De manière intéressante, des souris exposées à la perméthrine avant accouplement, présentent une descendance déficiente pour certaines fonctions comportementales (réflexe, nage…) (Farag et coll., 2006
). Certains pyréthrinoïdes perturbent la neuritogenèse
in vitro (PC12) à des concentrations micromolaires toutefois inférieures à celles provoquant une neurotoxicité chez l’adulte (Tran et coll., 2006
).
En résumé, l’impact des pyréthrinoïdes (perméthrine) sur le SNC et en particulier en période de développement est suggéré.
Troubles cognitifs et de l’humeur : lien avec les pesticides
La sérotonine (5HT) est un neurotransmetteur qui joue un rôle fondamental au niveau central dans le contrôle de l’appétit et de l’humeur. Les dérégulations du système sérotoninergique ont été associées à différentes pathologies comme les dépressions ou les dérèglements de l’appétit qui représentent des problématiques majeures en santé publique. Plusieurs études épidémiologiques suggèrent un lien entre exposition à certains types de pesticides et ces pathologies.
Organophosphorés
Les organophosphorés (OPs) ont, à ce titre, fait l’objet d’une attention particulière et au début des années 2000, des études expérimentales démontrent que l’exposition de modèles animaux (principalement des rats) à du chlorpyrifos modifie de manière significative, l’expression des récepteurs sérotoninergiques (5HTR) sur différentes cibles (Aldridge et coll., 2003
). Plusieurs études récentes ont précisé ces observations.
Ainsi, l’exposition de rats Sprague-Dawley à des doses non toxiques de chlorpyrifos (5 mg/kg/jour, dose inférieure à celles nécessaires pour cibler l’acétylcholine estérase–AChE) pendant la période postnatale (entre le 11
e et 14
e jour suivant la naissance, PN11-14) et sacrifiés à PN45 montre que cet OP perturbe modérément (variation maximum de 30 %) l’expression de plusieurs 5HTR et du transporteur sérotoninergiques (5HTT) dans certaines régions (mésencéphale, tronc cérébral, hippocampe). Les effets observés sont pour la plupart, des augmentations. Une perturbation de la signalisation des 5HTR est également observée (baisse de l’activation des adénylates cyclases couplées aux récepteurs) avec un effet plus prononcé chez les mâles. Bien que modérés, ces effets (liés à une exposition pendant une période sensible du développement, la période périnatale) pourraient être à l’origine de troubles de l’appétit ou de l’humeur pendant la vie adolescente ou adulte (Aldridge et coll., 2005a
). Ainsi, la même équipe dans la continuité de cette étude, montre que des doses de 1 mg/kg/jour de chlorpyrifos pendant une période plus précoce (PN1-4) conduisent à des déficits comportementaux (modèles de dépression) chez l’adulte associés traditionnellement aux voies sérotoninergiques (l’utilisation d’un inhibiteur de récepteur 5HT2, la kétansérine, confirme cette présomption) (Aldridge et coll., 2005b
). Sur le plan mécanistique, cette même équipe démontre que les périodes prénatales et postnatales sont des fenêtres d’exposition critique avec une augmentation du métabolisme de la sérotonine qui semble plus spécifiquement affectée que d’autres neurotransmetteurs comme la dopamine (Aldridge et coll., 2005c
). L’effet du chlorpyrifos ne doit toutefois pas être généralisé à tous les OPs et soulève de ce fait, des questions quant aux conséquences d’une exposition à des mélanges d’OPs sur ce système sérotoninergique. Ainsi, en fonction de la molécule utilisée, Slotkin et coll. (2008c
) montrent que deux OP différents (chlorpyrifos et diazinon) augmentent tous deux l’expression de la tryptophane hydroxylase (enzyme qui limite la synthèse de sérotonine) à des degrés variés et diminuent l’expression des transporteurs sur un modèle cellulaire de phéochromocytome de rat (PC12) (Slotkin et coll., 2008c
). Cependant, leurs effets sont opposés en ce qui concerne les récepteurs (augmentés par le chlorpyrifos et diminués par le diazinon). Ces études sont particulièrement intéressantes car réalisées à des doses inférieures au seuil d’inhibition de l’AChE ou de celles qui conduisent à des défauts de croissance (seuil toxique). Elles suggèrent ainsi un mécanisme indépendant de l’AChE, cible traditionnelle évoquée sur le plan mécanistique, des OP. Certaines de ces études réalisées à hautes doses retrouvent toutefois des effets similaires à celles réalisées à plus basse dose (Moreno et coll., 2008
).
En résumé, l’impact des OP (chlorpyrifos) sur le développement de troubles de l’appétit ou de l’humeur à l’adolescence ou l’âge adulte après une exposition pendant la vie fœtale est suggéré. Le mécanisme d’action serait une perturbation du système sérotoninergique indépendante de l’AChE.
Organochlorés
Les effets différentiels des OP (précédemment présentés) peuvent être étendus à d’autres catégories de pesticides comme les organochlorés (désormais interdits). Sur le modèle de lignée cellulaire PC12, la dieldrine exerce un effet inducteur sur l’enzyme de synthèse (tryptophane hydroxylase) et agit tout comme le diazinon (OP) en diminuant l’expression des récepteurs (Slotkin et coll., 2008c
). L’utilisation de modèles animaux a permis de montrer un effet de certains OC sur le système sérotoninergique (Cabaleiro et coll., 2008
) : une exposition
in utero et pendant la lactation à de l’endosulfan (0,6 ou 6 mg/kg), chez des rats mâles (Sprague-Dawley) sacrifiés à différentes périodes (PND15, 30 et 60), montre une augmentation spécifique des niveaux de sérotonine (entre 25 et 40 %) sans modification des niveaux de dopamine et de noradrénaline. Tout comme les OPs, les effets dépendent du type de pesticide et ne peuvent être généralisés à une classe particulière. Ainsi, une autre étude utilisant le méthoxychlore montre des effets contrastés en fonction de la localisation cérébrale quant à la quantité de 5HT (Lafuente et coll., 2008
).
En résumé, l’impact des OC sur le système sérotoninergique est dépendante du type de molécule. Après exposition in utero et pendant la lactation à l’endosulfan, une augmentation de sérotonine est observée chez des rats mâles.
Roténone
L’augmentation des niveaux de sérotonine au niveau cérébral pourrait apparaître paradoxale dans la mise en place de pathologies associées à des dérégulations du système sérotoninergique mais il faut garder en mémoire que les dérégulations sont associées à la fois à des perturbations de l’expression des membres du système sérotoninergique (enzymes du métabolisme, récepteurs, transporteurs) mais aussi à leur localisation et à celle de la sérotonine (vésicules, cytoplasme, fente synaptique). Ainsi, une étude utilisant la roténone, traditionnellement utilisée dans le cadre des modèles induits de Parkinson, révèle que celle-ci peut également exercer une cytotoxicité sur des neurones sérotoninergiques (Ren et Feng, 2007
). Sur le plan mécanistique, la roténone agit comme la colchicine, un agent dépolymérisant les microtubules ce qui a pour conséquence une accumulation de vésicules dans les neurones sérotoninergiques. L’hypothèse des auteurs de l’étude pour expliquer le lien entre accumulation vésiculaire et mort cellulaire, serait le passage de la sérotonine dans le cytoplasme. Son excès de concentration vésiculaire augmenterait le gradient vésiculo-cytoplasmique, ce qui entraînerait une fuite vers le cytoplasme. Ce passage conduirait à une augmentation du catabolisme de la sérotonine qui produit des espèces réactives de l’oxygène (ERO) à l’origine de la mort cellulaire.
En résumé, la perturbation du système sérotoninergique pourrait également être impliquée dans l’effet de la roténone sur le SNC.
En conclusion, les pathologies du système nerveux central ou périphérique ont été identifiées et étudiées bien avant l’apparition des pesticides. Ceux-ci sont toutefois suspectés de favoriser l’apparition de certaines d’entre elles notamment la maladie de Parkinson et certains défauts neurocomportementaux. En effet, en plus de l’apport majeur des études épidémiologiques, un certain nombre de ces substances présente des propriétés compatibles avec un rôle incident dans le développement de ces pathologies. À titre d’exemple, de nombreux pesticides ciblent la mitochondrie et semblent en mesure de provoquer à la fois une augmentation de la concentration de dérivés réactifs de l’oxygène et une baisse de production d’ATP qui est fondamental pour la survie des neurones. Compte tenu des caractéristiques particulières des cellules dopaminergiques vis-à-vis du stress oxydant, des modèles de voies de signalisation peuvent ainsi être proposés pour expliquer les rôles potentiels des pesticides dans le développement de la maladie de Parkinson.
Les études expérimentales menées avec des modèles cellulaires ou animaux posent pour la plupart, des problèmes récurrents comme celui des hautes concentrations et des courtes périodes de traitement, très différentes des conditions d’exposition chez l’Homme (tout comme la voie d’exposition). À titre d’exemple, la plupart des études menées sur des modèles animaux murins, utilisent des concentrations de pesticides proches de 1 mg/kg sur de courtes périodes alors que les valeurs de référence pour la population humaine sont beaucoup plus basses (par exemple : dose journalière maximale tolérée, 5 µg/kg) et généralement respectées (WHO, 2008) même chez les agriculteurs exposés (<1 µg/kg/j ; Lee et coll., 2009).
Dans le cadre de la maladie de Parkinson, les rares protocoles utilisant les voies d’exposition les plus classiques chez l’Homme (derme, système respiratoire) n’ont pas permis de démontrer un effet sur la substance noire (Rojo et coll., 2007
). Des études menées à l’aide d’échantillons humains ont toutefois permis de montrer que de basses concentrations sanguines de pesticides pouvaient être associées à une hypométhylation de l’ADN (et avoir un effet potentiel sur l’expression des gènes) (Kim et coll., 2010
). Ces nouvelles données posent le problème de la transmission de l’information entre générations et ce, pour l’ensemble des tissus de l’organisme dont le système nerveux. Il est également possible que les pesticides puissent avoir un effet très différent à hautes et basses concentrations (effet type courbe en U) comme cela a été démontré pour d’autres perturbateurs endocriniens comme le bisphénol A, posant la question de la légitimité d’un grand nombre d’études expérimentales. Néanmoins, pour une pathologie comme la maladie de Parkinson, l’identification de marqueurs du stress oxydant dans la substance noire d’individus (post-mortem) légitime quelque peu les modèles de voies de signalisation activées par des pesticides comme le paraquat.
En plus des concentrations, le temps et la période d’exposition sont des paramètres également importants à prendre en compte dans les analyses. Il est fortement probable que chez un patient développant une maladie de Parkinson, la substance noire ait été agressée à différentes périodes du développement de l’individu conduisant à une sensibilisation progressive du tissu et à sa dégénérescence (
the multi-hit hypothesis, Barlow et coll., 2007
). Cette hypothèse cadre bien avec le mode d’exposition des travailleurs agricoles qui sont exposés la plupart du temps à la fois à plusieurs produits et à intervalles réguliers. On peut ainsi imaginer que certaines cellules présentent des signes d’apoptose « inachevée » (abortose), préambule aux développements pathologiques marqués par les manifestations phénotypiques. La fenêtre d’exposition est un paramètre important à considérer notamment pour les jeunes organismes (embryon, fœtus, jeune enfant en tenant compte du rôle de la barrière placentaire) qui seraient potentiellement plus sensibles car en cours de développement. Toutefois, cette notion peut être modulée car, par exemple dans le cadre d’une intoxication aiguë aux organophosphates, la protéine cible (acétylcholine estérase) est plus rapidement re-synthétisée chez les plus jeunes individus qui seraient donc capables de récupérer plus vite. Ce constat reste compatible avec des effets chroniques plus néfastes (Thiruchelvam et coll., 2002
).
Comme évoqué précédemment dans le cadre de l’exposition des travailleurs agricoles, les études utilisant des expositions multiples sont particulièrement intéressantes à considérer (exemple du manèbe et du paraquat) et sont encore rares (Bjorling-Poulsen et coll., 2008
; Thiruchelvam et coll., 2000
). Au-delà des mélanges n’impliquant que des pesticides, l’étude de l’influence d’autres polluants environnementaux voire même de médicaments (ciblant par exemple les voies de l’inflammation compte tenu des données évoquées sur les cellules gliales ou perturbant des processus importants comme le transport membranaire) sur l’action de ces pesticides semble également judicieuse notamment quand ceux-ci sont des xénobiotiques persistants (cas des
PolyChlorinated Biphenyls ou PCBs dans la maladie de Parkinson).
Par ailleurs, les pesticides pourraient agir indirectement sur le système nerveux. À titre d’exemple, le produit de dégradation des EBDC, l’éthylènethiourée (ETU), est un inhibiteur de la synthèse des hormones thyroïdes qui joue un rôle important dans la mise en place du système nerveux et de son homéostasie (Doerge et Takazawa 1990
; Marinovich et coll., 1997
). De nombreux pesticides lient, par ailleurs, plusieurs récepteurs nucléaires (récepteurs aux œstrogènes, aux androgènes, PXR, CAR) qui modulent l’expression de métabolismes endogènes ou de xénobiotiques. Ce phénomène de perturbation endocrinienne pourrait conduire à une augmentation de la concentration de certains neurotoxiques (non pesticides) d’origine naturelle (dont ceux de la dopamine) ou anthropique (Le Couteur et coll., 1999
). Dans le même registre, des études récentes suggèrent que certains pesticides (organophosphorés, roténone, pyréthrinoïdes, organochlorés, paraquat) modifient les concentrations de sérotonine qui joue un rôle important dans la régulation de l’humeur.
Enfin, les pesticides pourraient constituer des marqueurs d’autres expositions à des neurotoxiques. De nombreux pesticides sont par exemple, dissous dans des solvants qui pourraient exercer une neurotoxicité. Par ailleurs, certains auteurs montrent que des neurotoxines (comme l’époxomicine d’origine microbienne) induisent des syndromes parkinsoniens chez le rat par inhibition du système de dégradation dépendant du protéasome (McNaught et coll., 2004
; Caldwell et coll., 2009
). Néanmoins, ces effets ne se retrouvent pas avec tous les inhibiteurs du protéasome (Kordower et coll., 2006
).
Les nombreuses données produites par les premières études expérimentales sur l’effet des pesticides à hautes doses ont permis d’identifier un certain nombre de mécanismes d’action de ces composés qui semblent compatibles avec les données physiopathologiques des maladies auxquelles ils sont associés par les études épidémiologiques. Il faut désormais encourager également les études sur de longues périodes utilisant des mélanges à basses concentrations ciblant une ou plusieurs voies de signalisation pour renforcer ces premières hypothèses mécanistiques et identifier des marqueurs précoces des pathologies dans un but préventif. Les modèles animaux représentent des outils attractifs pour mener ces études d’autant qu’ils permettent d’analyser plus facilement les interactions entre génétique et environnement. Le rôle de certains gènes dans le développement de pathologies comme la maladie de Parkinson (par exemple : PINK1, Parkine, DJ-1), renforce d’ailleurs certains modèles d’action des pesticides de par la convergence de certains mécanismes d’action (par exemple : dysfonctionnement mitochondrial). Le rôle des polymorphismes des enzymes du métabolisme pourrait être non négligeable (par exemple : paraoxonase 1). À l’heure actuelle, aucune étude expérimentale menée sur les pesticides n’a toutefois permis de retrouver sur les modèles animaux utilisés toutes les caractéristiques de la maladie de Parkinson (Cicchetti et coll., 2009
). Compte tenu également des divergences entre Homme et animaux, les modèles cellulaires de par leur facilité d’action, seront donc toujours des outils complémentaires importants dans ces études.
Bibliographie
[1] ADAMS J, VORHEES CV, MIDDAUGH LD. Developmental neurotoxicity of anticonvulsants: human and animal evidence on phenytoin.
Neurotoxicol Teratol. 1990;
12:203
-214

[2] ADIGUN AA, RYDE IT, SEIDLER FJ, SLOTKIN TA. Organophosphate exposure during a critical developmental stage reprograms adenylyl cyclase signaling in PC12 cells.
Brain Res. 2010a;
1329:36
-44
[3] ADIGUN AA, SEIDLER FJ, SLOTKIN TA, . Disparate developmental neurotoxicants converge on the cyclic AMP signaling cascade, revealed by transcriptional profiles in vitro and in vivo.
Brain Res. 2010b;
1316:1
-16
[4] AHLBOM J, FREDRIKSSON A, ERIKSSON P. Neonatal exposure to a type-I pyrethroid (bioallethrin) induces dose-response changes in brain muscarinic receptors and behaviour in neonatal and adult mice.
Brain Res. 1994;
645:318
-324
[5] AHMADI FA, GRAMMATOPOULOS TN, POCZOBUTT AM, JONES SM, SNELL LD, et coll. Dopamine selectively sensitizes dopaminergic neurons to rotenone-induced apoptosis.
Neurochem Res. 2008;
33:886
-901
[6] AHMADI FA, LINSEMAN DA, GRAMMATOPOULOS TN, JONES SM, BOUCHARD RJ, et coll. The pesticide rotenone induces caspase-3-mediated apoptosis in ventral mesencephalic dopaminergic neurons.
J Neurochem. 2003;
87:914
-921
[7] ALAM M, SCHMIDT WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats.
Behav Brain Res. 2002;
136:317
-324
[8] ALDRIDGE JE, LEVIN ED, SEIDLER FJ, SLOTKIN TA. Developmental exposure of rats to chlorpyrifos leads to behavioral alterations in adulthood, involving serotonergic mechanisms and resembling animal models of depression.
Environ Health Perspect. 2005a;
113:527
-531
[9] ALDRIDGE JE, MEYER A, SEIDLER FJ, SLOTKIN TA. Developmental exposure to terbutaline and chlorpyrifos: pharmacotherapy of preterm labor and an environmental neurotoxicant converge on serotonergic systems in neonatal rat brain regions.
Toxicol Appl Pharmacol. 2005b;
203:132
-144
[10] ALDRIDGE JE, MEYER A, SEIDLER FJ, SLOTKIN TA. Alterations in central nervous system serotonergic and dopaminergic synaptic activity in adulthood after prenatal or neonatal chlorpyrifos exposure.
Environ Health Perspect. 2005c;
113:1027
-1031
[11] ALDRIDGE JE, SEIDLER FJ, MEYER A, THILLAI I, SLOTKIN TA. Serotonergic systems targeted by developmental exposure to chlorpyrifos: effects during different critical periods.
Environ Health Perspect. 2003;
111:1736
-1743
[12] ANANTHARAM V, KITAZAWA M, WAGNER J, KAUL S, KANTHASAMY AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl.
J Neurosci. 2002;
22:1738
-1751
[13] ANDRES-MATEOS E, MEJIAS R, SASAKI M, LI X, LIN BM, et coll. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine).
J Neurosci. 2009;
29:15846
-15850
[14] ASCHERIO A, ZHANG SM, HERNAN MA, KAWACHI I, COLDITZ GA, et coll. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women.
Ann Neurol. 2001;
50:56
-63
[15] AZIZ MH, AGRAWAL AK, ADHAMI VM, SHUKLA Y, SETH PK. Neurodevelopmental consequences of gestational exposure (GD14-GD20) to low dose deltamethrin in rats.
Neurosci Lett. 2001;
300:161
-165
[16] BACHURIN SO, SHEVTZOVA EP, LERMONTOVA NN, SERKOVA TP, RAMSAY RR. The effect of dithiocarbamates on neurotoxic action of 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) and on mitochondrial respiration chain.
Neurotoxicology. 1996;
17:897
-903
[17] BAGETTA G, CORASANITI MT, IANNONE M, NISTICO G, STEPHENSON JD. Production of limbic motor seizures and brain damage by systemic and intracerebral injections of paraquat in rats.
Pharmacol Toxicol. 1992;
71:443
-448
[18] BAKER DB, LOO S, BARKER J. Evaluation of human exposure to the heptachlor epoxide contamination of milk in Hawaii.
Hawaii Med J. 1991;
50:108-12
-118
[19] BANERJEE BD, SETH V, AHMED RS. Pesticide-induced oxidative stress: perspectives and trends.
Rev Environ Health. 2001;
16:1
-40
[20] BARLOW BK, CORY-SLECHTA DA, RICHFIELD EK, THIRUCHELVAM M. The gestational environment and Parkinson’s disease: evidence for neurodevelopmental origins of a neurodegenerative disorder.
Reprod Toxicol. 2007;
23:457
-470
[21] BARLOW BK, LEE DW, CORY-SLECHTA DA, OPANASHUK LA. Modulation of antioxidant defense systems by the environmental pesticide maneb in dopaminergic cells.
Neurotoxicology. 2005;
26:63
-75
[22] BARLOW BK, THIRUCHELVAM MJ, BENNICE L, CORY-SLECHTA DA, BALLATORI N, et coll. Increased synaptosomal dopamine content and brain concentration of paraquat produced by selective dithiocarbamates.
J Neurochem. 2003;
85:1075
-1086
[23] BARTELS AL, KORTEKAAS R, BART J, WILLEMSEN AT, DE KLERK OL, et coll. Blood--brain barrier P-glycoprotein function decreases in specific brain regions with aging: a possible role in progressive neurodegeneration.
Neurobiol Aging. 2009;
30:1818
-1824
[24] BARTLETT RM, HOLDEN JE, NICKLES RJ, MURALI D, BARBEE DL, et coll. Paraquat is excluded by the blood brain barrier in rhesus macaque: An in vivo pet study.
Brain Res. 2009;
1259:74
-79
[25] BARTLETT RM, MURALI D, NICKLES RJ, BARNHART TE, HOLDEN JE, et coll. Assessment of Fetal Brain Uptake of Paraquat In Utero Using In Vivo PET/CT Imaging.
Toxicol Sci. 2011;
122:551
-556
[26] BASHKATOVA V, ALAM M, VANIN A, SCHMIDT WJ. Chronic administration of rotenone increases levels of nitric oxide and lipid peroxidation products in rat brain.
Exp Neurol. 2004;
186:235
-241
[27] BERGEN WG. The in vitro effect of dieldrin on respiration of rat liver mitochondria.
Proc Soc Exp Biol Med. 1971;
136:732
-735
[28] BETARBET R, SHERER TB, DI MONTE DA, GREENAMYRE JT. Mechanistic approaches to Parkinson’s disease pathogenesis.
Brain Pathol. 2002;
12:499
-510
[29] BETARBET R, SHERER TB, MACKENZIE G, GARCIA-OSUNA M, PANOV AV, et coll. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease.
Nat Neurosci. 2000;
3:1301
-1306
[30] BHARATH S, HSU M, KAUR D, RAJAGOPALAN S, ANDERSEN JK. Glutathione, iron and Parkinson’s disease.
Biochem Pharmacol. 2002;
64:1037
-1048
[31] BJORLING-POULSEN M, ANDERSEN HR, GRANDJEAN P. Potential developmental neurotoxicity of pesticides used in Europe.
Environ Health. 2008;
7:50
[32] BLOOMQUIST JR, BARLOW RL, GILLETTE JS, LI W. KIRBY ML. Selective effects of insecticides on nigrostriatal dopaminergic nerve pathways.
Neurotoxicology. 2002;
23:537
-544
[33] BOMSER J, CASIDA JE. Activation of extracellular signal-regulated kinases (ERK 44/42) by chlorpyrifos oxon in Chinese hamster ovary cells.
J Biochem Mol Toxicol. 2000;
14:346
-353
[34] BOMSER JA, QUISTAD GB, CASIDA JE. Chlorpyrifos oxon potentiates diacylglycerol-induced extracellular signal-regulated kinase (ERK 44/42) activation, possibly by diacylglycerol lipase inhibition.
Toxicol Appl Pharmacol. 2002;
178:29
-36
[35] BONGIOVANNI B, DE LORENZI P, FERRI A, KONJUH C, RASSETTO M, et coll. Melatonin decreases the oxidative stress produced by 2,4-dichlorophenoxyacetic acid in rat cerebellar granule cells.
Neurotox Res. 2007;
11:93
-99
[36] BORTOLOZZI A, EVANGELISTA DE DUFFARD AM, DAJAS F,, DUFFARD R, SILVEIRA R. Intracerebral administration of 2,4-diclorophenoxyacetic acid induces behavioral and neurochemical alterations in the rat brain.
Neurotoxicology. 2001;
22:221
-232
[37] BORTOLOZZI AA, DUFFARD RO, EVANGELISTA DE DUFFARD AM. Behavioral alterations induced in rats by a pre- and postnatal exposure to 2,4-dichlorophenoxyacetic acid.
Neurotoxicol Teratol. 1999;
21:451
-465
[38] BORTOLOZZI AA, EVANGELISTA DE DUFFARD AM, DUFFARD RO, ANTONELLI MC. Effects of 2,4-dichlorophenoxyacetic acid exposure on dopamine D2-like receptors in rat brain.
Neurotoxicol Teratol. 2004;
26:599
-605
[39] BOWER JH, MARAGANORE DM, PETERSON BJ, MCDONNELL SK, AHLSKOG JE, et coll. Head trauma preceding PD: a case-control study.
Neurology. 2003;
60:1610
-1615
[40] BRADBERRY SM, CAGE SA, PROUDFOOT AT, VALE JA. Poisoning due to pyrethroids.
Toxicol Rev. 2005;
24:93
-106
[41] BRADBERRY SM, WATT BE, PROUDFOOT AT, VALE JA. Mechanisms of toxicity, clinical features, and management of acute chlorophenoxy herbicide poisoning: a review.
J Toxicol Clin Toxicol. 2000;
38:111
-122
[42] BROOKS AI, CHADWICK CA, GELBARD HA, CORY-SLECHTA DA, FEDEROFF HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss.
Brain Res. 1999;
823:1
-10
[43] BROPHY VH, HASTINGS MD, CLENDENNING JB, RICHTER RJ, JARVIK GP, et coll. Polymorphisms in the human paraoxonase (PON1) promoter.
Pharmacogenetics. 2001;
11:77
-84
[44] CABALEIRO T, CARIDE A, ROMERO A, LAFUENTE A. Effects of in utero and lactational exposure to endosulfan in prefrontal cortex of male rats.
Toxicol Lett. 2008;
176:58
-67
[45] CABONI P, SHERER TB, ZHANG N, TAYLOR G, NA HM, et coll. Rotenone, deguelin, their metabolites, and the rat model of Parkinson’s disease.
Chem Res Toxicol. 2004;
17:1540
-1548
[46] CALDWELL KA, TUCCI ML, ARMAGOST J, HODGES TW, CHEN J, et coll. Investigating bacterial sources of toxicity as an environmental contributor to dopaminergic neurodegeneration.
PLoS One. 2009;
4:e7227
[47] CALO M, IANNONE M, PASSAFARO M, NISTICO G. Selective vulnerability of hippocampal CA3 neurones after microinfusion of paraquat into the rat substantia nigra or into the ventral tegmental area.
J Comp Pathol. 1990;
103:73
-78
[48] CAMPBELL CG, SEIDLER FJ, SLOTKIN TA. Chlorpyrifos interferes with cell development in rat brain regions.
Brain Res Bull. 1997;
43:179
-189
[49] CANET-AVILES RM, WILSON MA, MILLER DW, AHMAD R, MCLENDON C, et coll. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization.
Proc Natl Acad Sci USA. 2004;
101:9103
-9108
[50] CANTALAMESSA F. Acute toxicity of two pyrethroids, permethrin, and cypermethrin in neonatal and adult rats.
Arch Toxicol. 1993;
67:510
-513
[51] CASAREJOS MJ, MENENDEZ J, SOLANO RM, RODRIGUEZ-NAVARRO JA, GARCIA DE YEBENES J, et coll. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline.
J Neurochem. 2006;
97:934
-946
[52] CASTELLO PR, DRECHSEL DA, PATEL M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain.
J Biol Chem. 2007;
282:14186
-14193
[53] CAUGHLAN A, NEWHOUSE K, NAMGUNG U, XIA Z. Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases.
Toxicol Sci. 2004;
78:125
-134
[54] CERSOSIMO MG, KOLLER WC. The diagnosis of manganese-induced parkinsonism.
Neurotoxicology. 2006;
27:340
-346
[55] CHAN MP, MORISAWA S, NAKAYAMA A, KAWAMOTO Y, SUGIMOTO M, et coll. A physiologically based pharmacokinetic model for endosulfan in the male Sprague-Dawley rats.
Environ Toxicol. 2006a;
21:464
-478
[56] CHAN MP, MORISAWA S, NAKAYAMA A, KAWAMOTO Y, YONEDA M. Development of an in vitro blood-brain barrier model to study the effects of endosulfan on the permeability of tight junctions and a comparative study of the cytotoxic effects of endosulfan on rat and human glial and neuronal cell cultures.
Environ Toxicol. 2006;
21:223
-235
[57] CHANYACHUKUL T, YOOVATHAWORN K, THONGSAARD W, CHONGTHAMMAKUN S, NAVASUMRIT P, et coll. Attenuation of paraquat-induced motor behavior and neurochemical disturbances by L-valine in vivo.
Toxicol Lett. 2004;
150:259
-269
[58] CHIBA-FALEK O, NUSSBAUM RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system.
Hum Mol Genet. 2001;
10:3101
-3109
[59] CHOI HS, AN JJ, KIM SY, LEE SH, KIM DW, et coll. PEP-1-SOD fusion protein efficiently protects against paraquat-induced dopaminergic neuron damage in a Parkinson disease mouse model.
Free Radic Biol Med. 2006;
41:1058
-1068
[60] CHOI WS, KRUSE SE, PALMITER RD, XIA Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat.
Proc Natl Acad Sci USA. 2008;
105:15136
-15141
[61] CHOU AP, MAIDMENT N, KLINTENBERG R, CASIDA JE, LI S, et coll. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome.
J Biol Chem. 2008;
283:34696
-34703
[62] CHUN HS, GIBSON GE, DEGIORGIO LA, ZHANG H, KIDD VJ, et coll. Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism.
J Neurochem. 2001;
76:1010
-1021
[63] CICCHETTI F, DROUIN-OUELLET J, GROSS RE. Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models?.
Trends Pharmacol Sci. 2009;
30:475
-483
[64] CICCHETTI F, LAPOINTE N, ROBERGE-TREMBLAY A, SAINT-PIERRE M, JIMENEZ L, et coll. Systemic exposure to paraquat and maneb models early Parkinson’s disease in young adult rats.
Neurobiol Dis. 2005;
20:360
-371
[65] CLAYTON R, CLARK JB, SHARPE M. Cytochrome c release from rat brain mitochondria is proportional to the mitochondrial functional deficit: implications for apoptosis and neurodegenerative disease.
J Neurochem. 2005;
92:840
-849
[66] COCHEME HM, MURPHY MP. Complex I is the major site of mitochondrial superoxide production by paraquat.
J Biol Chem. 2008;
283:1786
-1798
[67] COLE TB, WALTER BJ, SHIH DM, TWARD AD, LUSIS AJ, et coll. Toxicity of chlorpyrifos and chlorpyrifos oxon in a transgenic mouse model of the human paraoxonase (PON1) Q192R polymorphism.
Pharmacogenet Genomics. 2005;
15:589
-598
[68] COLE TB, BEYER RP, BAMMLER TK, PARK SS, FARIN FM, et coll. Repeated Developmental Exposure of Mice to Chlorpyrifos Oxon Is Associated with Paraoxonase 1 (PON1)-Modulated Effects on Cerebellar Gene Expression.
Toxicol Sci. 2011;
123:155
-169
[69] CORASANITI MT, STRONGOLI MC, ROTIROTI D, BAGETTA G, NISTICO G. Paraquat: a useful tool for the in vivo study of mechanisms of neuronal cell death.
Pharmacol Toxicol. 1998;
83:1
-7
[70] CORRIGAN FM, MURRAY L, WYATT CL, SHORE RF. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson’s disease.
Exp Neurol. 1998;
150:339
-342
[71] CORRIGAN FM, WIENBURG CL, SHORE RF, DANIEL SE, MANN D. Organochlorine insecticides in substantia nigra in Parkinson’s disease.
J Toxicol Environ Health A. 2000;
59:229
-234
[72] CORY-SLECHTA DA, THIRUCHELVAM M, RICHFIELD EK, BARLOW BK, BROOKS AI. Developmental pesticide exposures and the Parkinson’s disease phenotype.
Birth Defects Res A Clin Mol Teratol. 2005;
73:136
-139
[73] COSTA LG, RICHTER RJ, LI WF, COLE T, GUIZZETTI M, et coll. Paraoxonase (PON 1) as a biomarker of susceptibility for organophosphate toxicity.
Biomarkers. 2003;
8:1
-12
[74] COULOM H, BIRMAN S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster.
J Neurosci. 2004;
24:10993
-10998
[75] CRISTOVAO AC, CHOI DH, BALTAZAR G, BEAL MF, KIM YS. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death.
Antioxid Redox Signal. 2009;
11:2105
-2118
[76] CRUMPTON TL, SEIDLER FJ, SLOTKIN TA. Developmental neurotoxicity of chlorpyrifos in vivo and in vitro: effects on nuclear transcription factors involved in cell replication and differentiation.
Brain Res. 2000;
857:87
-98
[77] DAM K, SEIDLER FJ, SLOTKIN TA. Chlorpyrifos exposure during a critical neonatal period elicits gender-selective deficits in the development of coordination skills and locomotor activity.
Brain Res Dev Brain Res. 2000;
121:179
-187
[78] DAM K, SEIDLER FJ, SLOTKIN TA. Developmental neurotoxicity of chlorpyrifos: delayed targeting of DNA synthesis after repeated administration.
Brain Res Dev Brain Res. 1998;
108:39
-45
[79] DAM K, SEIDLER FJ, SLOTKIN TA. Transcriptional biomarkers distinguish between vulnerable periods for developmental neurotoxicity of chlorpyrifos: Implications for toxicogenomics.
Brain Res Bull. 2003;
59:261
-265
[80] DAUER W, KHOLODILOV N, VILA M, TRILLAT AC, GOODCHILD R, et coll. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP.
Proc Natl Acad Sci USA. 2002;
99:14524
-14529
[81] DAVIES HG, RICHTER RJ, KEIFER M, BROOMFIELD CA, SOWALLA J, et coll. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin.
Nat Genet. 1996;
14:334
-336
[82] DEBBARH I, MOORE N. A simple method for the determination of ethylene-thiourea (ETU) in biological samples.
J Anal Toxicol. 2002;
26:216
-221
[83] DENG H, JANKOVIC J, GUO Y, XIE W, LE W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y.
Biochem Biophys Res Commun. 2005;
337:1133
-1138
[84] DI MONTE DA, LAVASANI M, MANNING-BOG AB. Environmental factors in Parkinson’s disease.
Neurotoxicology. 2002;
23:487
-502
[85] DOERGE DR, TAKAZAWA RS. Mechanism of thyroid peroxidase inhibition by ethylenethiourea.
Chem Res Toxicol. 1990;
3:98
-101
[86] DOMICO LM, ZEEVALK GD, BERNARD LP, COOPER KR. Acute neurotoxic effects of mancozeb and maneb in mesencephalic neuronal cultures are associated with mitochondrial dysfunction.
Neurotoxicology. 2006;
27:816
-825
[87] DRECHSEL DA, PATEL M. Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism.
Toxicol Sci. 2009;
112:427
-434
[88] DROZDZIK M, BIALECKA M, MYSLIWIEC K, HONCZARENKO K, STANKIEWICZ J, SYCH Z. Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease.
Pharmacogenetics. 2003;
13:259
-263
[89] DUFFARD R, GARCIA G, ROSSO S, BORTOLOZZI A, MADARIAGA M, et coll. Central nervous system myelin deficit in rats exposed to 2,4-dichlorophenoxyacetic acid throughout lactation.
Neurotoxicol Teratol. 1996;
18:691
-696
[90] DUTHEIL F, BEAUNE P, LORIOT MA, . Xenobiotic metabolizing enzymes in the central nervous system: Contribution of cytochrome P450 enzymes in normal and pathological human brain.
Biochimie. 2008;
90:426
-436
[91] DUTHEIL F, BEAUNE P, TZOURIO C, LORIOT MA, ELBAZ A. Interaction between ABCB1 and professional exposure to organochlorine insecticides in Parkinson disease.
Arch Neurol. 2010;
67:739
-45
[92] ELBAZ A, LEVECQUE C, CLAVEL J, VIDAL JS, RICHARD F, et coll. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease.
Ann Neurol. 2004;
55:430
-434
[93] ELBAZ A, MOISAN F. Update in the epidemiology of Parkinson’s disease.
Curr Opin Neurol. 2008;
21:454
-460
[94] ELO HA, MACDONALD E. Effects of 2,4-dichlorophenoxyacetic acid (2,4-D) on biogenic amines and their acidic metabolites in brain and cerebrospinal fluid of rats.
Arch Toxicol. 1989;
63:127
-130
[95] ELO HA, YLITALO P. Distribution of 2-methyl-4-chlorophenoxyacetic acid and 2,4-dichlorophenoxyacetic acid in male rats: evidence for the involvement of the central nervous system in their toxicity.
Toxicol Appl Pharmacol. 1979;
51:439
-446
[96] ELWAN MA, RICHARDSON JR, GUILLOT TS, CAUDLE WM, MILLER GW. Pyrethroid pesticide-induced alterations in dopamine transporter function.
Toxicol Appl Pharmacol. 2006;
211:188
-197
[97] ERIKSSON P, FREDRIKSSON A. Neurotoxic effects of two different pyrethroids, bioallethrin and deltamethrin, on immature and adult mice: changes in behavioral and muscarinic receptor variables.
Toxicol Appl Pharmacol. 1991;
108:78
-85
[98] ERIKSSON P, NORDBERG A. Effects of two pyrethroids, bioallethrin and deltamethrin, on subpopulations of muscarinic and nicotinic receptors in the neonatal mouse brain.
Toxicol Appl Pharmacol. 1990;
102:456
-463
[99] FARAG AT, GODA NF, MANSEE AH, SHAABAN NA. Effects of permethrin given before mating on the behavior of F1-generation in mice.
Neurotoxicology. 2006;
27:421
-428
[100] FEI Q, ETHELL DW. Maneb potentiates paraquat neurotoxicity by inducing key Bcl-2 family members.
J Neurochem. 2008;
105:2091
-2097
[101] FERNAGUT PO, HUTSON CB, FLEMING SM, TETREAUT NA, SALCEDO J, et coll. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression.
Synapse. 2007;
61:991
-1001
[102] FERRANTE RJ, SCHULZ JB, KOWALL NW, BEAL MF. Systemic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra.
Brain Res. 1997;
753:157
-162
[103] FERRAZ HB, BERTOLUCCI PH, PEREIRA JS, LIMA JG, ANDRADE LA. Chronic exposure to the fungicide maneb may produce symptoms and signs of CNS manganese intoxication.
Neurology. 1988;
38:550
-553
[104] FERRI A, DUFFARD R, DE DUFFARD AM. Selective oxidative stress in brain areas of neonate rats exposed to 2,4-Dichlorophenoxyacetic acid through mother’s milk.
Drug Chem Toxicol. 2007;
30:17
-30
[105] FITSANAKIS VA, AMARNATH V, MOORE JT, MONTINE KS, ZHANG J, et coll. Catalysis of catechol oxidation by metal-dithiocarbamate complexes in pesticides.
Free Radic Biol Med. 2002;
33:1714
-1723
[106] FLEMING L, MANN JB, BEAN J, BRIGGLE T, SANCHEZ-RAMOS JR. Parkinson’s disease and brain levels of organochlorine pesticides.
Ann Neurol. 1994;
36:100
-103
[107] FOLEY P, RIEDERER P. Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson’s disease.
J Neurol. 2000;
247 (suppl 2):II82
-II94
[108] FURLONG CE. Genetic variability in the cytochrome P450-paraoxonase 1 (PON1) pathway for detoxication of organophosphorus compounds.
J Biochem Mol Toxicol. 2007;
21:197
-205
[109] GAO HM, HONG JS, ZHANG W, LIU B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons.
J Neurosci. 2002;
22:782
-790
[110] GAO HM, LIU B, HONG JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons.
J Neurosci. 2003;
23:6181
-6187
[111] GARCIA SJ, SEIDLER FJ, CRUMPTON TL, SLOTKIN TA. Does the developmental neurotoxicity of chlorpyrifos involve glial targets? Macromolecule synthesis, adenylyl cyclase signaling, nuclear transcription factors, and formation of reactive oxygen in C6 glioma cells.
Brain Res. 2001;
891:54
-68
[112] GARCIA SJ, SEIDLER FJ, QIAO D, SLOTKIN TA. Chlorpyrifos targets developing glia: effects on glial fibrillary acidic protein.
Brain Res Dev Brain Res. 2002;
133:151
-161
[113] GELDMACHER DS. Stimulus characteristics determine processing approach on random array letter-cancellation tasks.
Brain Cogn. 1998;
36:346
-354
[114] GHERSI-EGEA JF, LIVERTOUX MH, MINN A, PERRIN R, SIEST G. Enzyme mediated superoxide radical formation initiated by exogenous molecules in rat brain preparations.
Toxicol Appl Pharmacol. 1991;
110:107
-117
[115] GILLETTE JS, BLOOMQUIST JR. Differential up-regulation of striatal dopamine transporter and alpha-synuclein by the pyrethroid insecticide permethrin.
Toxicol Appl Pharmacol. 2003;
192:287
-293
[116] GIMENEZ-CASSINA A, LIM F, CERRATO T, PALOMO GM, DIAZ-NIDO J. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3.
J Biol Chem. 2009;
284:3001
-3011
[117] GONZALEZ-POLO R, NISO-SANTANO M, MORAN JM, ORTIZ-ORTIZ MA, BRAVO-SAN PEDRO JM, et coll. Silencing DJ-1 reveals its contribution in paraquat-induced autophagy.
J Neurochem. 2009;
109:889
-898
[118] GONZALEZ-POLO RA, NISO-SANTANO M, ORTIZ-ORTIZ MA, GOMEZ-MARTIN A, MORAN JM, et coll. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells.
Toxicol Sci. 2007;
97:448
-458
[119] GONZALEZ-POLO RA, RODRIGUEZ-MARTIN A, MORAN JM, NISO M, SOLER G, et coll. Paraquat-induced apoptotic cell death in cerebellar granule cells.
Brain Res. 2004;
1011:170
-176
[120] GOUEDARD C, BAROUKI R, MOREL Y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism.
Mol Cell Biol. 2004;
24:5209
-5222
[121] GREENAMYRE JT, MACKENZIE G, PENG TI, STEPHANS SE. Mitochondrial dysfunction in Parkinson’s disease.
Biochem Soc Symp. 1999;
66:85
-97
[122] GREENAMYRE JT, SHERER TB, BETARBET R, PANOV AV. Complex I and Parkinson’s disease.
IUBMB Life. 2001;
52:135
-141
[123] GRUNDEMANN J, SCHLAUDRAFF F, HAECKEL O, LISS B. Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease.
Nucleic Acids Res. 2008;
36:e38
[124] GUPTA A, AGARWAL R, SHUKLA GS. Functional impairment of blood-brain barrier following pesticide exposure during early development in rats.
Hum Exp Toxicol. 1999;
18:174
-179
[125] GUPTA RC, MILATOVIC S, DETTBARN WD, ASCHNER M, MILATOVIC D. Neuronal oxidative injury and dendritic damage induced by carbofuran: protection by memantine.
Toxicol Appl Pharmacol. 2007;
219:97
-105
[126] GUPTA SP, PATEL S, YADAV S, SINGH AK, SINGH S, et coll. Involvement of nitric oxide in maneb- and paraquat-induced Parkinson’s disease phenotype in mouse: is there any link with lipid peroxidation?.
Neurochem Res. 2010;
35:1206
-1213
[127] HAQUE ME, THOMAS KJ, D’SOUZA C, CALLAGHAN S, KITADA T, et coll. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP.
Proc Natl Acad Sci USA. 2008;
105:1716
-1721
[128] HARGREAVES AJ. Neurodegenerations induced by organophosphorous compounds.
Adv Exp Med Biol. 2012;
724:189
-204
[129] HARTMANN A, HUNOT S, MICHEL PP, MURIEL MP, VYAS S, et coll. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease.
Proc Natl Acad Sci USA. 2000;
97:2875
-2880
[130] HATCHER JM, PENNELL KD, MILLER GW. Parkinson’s disease and pesticides: a toxicological perspective.
Trends Pharmacol Sci. 2008;
29(6):322
-329
[131] HATCHER JM, RICHARDSON JR, GUILLOT TS, MCCORMACK AL, DI MONTE DA, et coll. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system.
Exp Neurol. 2007;
204:619
-630
[132] HE Y, IMAM SZ, DONG Z, JANKOVIC J, ALI SF, et coll. Role of nitric oxide in rotenone-induced nigro-striatal injury.
J Neurochem. 2003;
86:1338
-1345
[133] HEATH PR, SHAW PJ. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis.
Muscle Nerve. 2002;
26(4):438
-458
[134] HEINZ GH, HILL EF, CONTRERA JF. Dopamine and norepinephrine depletion in ring doves fed DDE, dieldrin, and Aroclor 1254.
Toxicol Appl Pharmacol. 1980;
53:75
-82
[135] HERNANDEZ AF, MACKNESS B, RODRIGO L, LOPEZ O, PLA A, et coll. Paraoxonase activity and genetic polymorphisms in greenhouse workers with long term pesticide exposure.
Hum Exp Toxicol. 2003;
22:565
-574
[136] HERVONEN H, ELO HA, YLITALO P. Blood-brain barrier damage by 2-methyl-4-chlorophenoxyacetic acid herbicide in rats.
Toxicol Appl Pharmacol. 1982;
65:23
-31
[137] HOCHMAN A, STERNIN H, GORODIN S, KORSMEYER S, ZIV I, et coll. Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice.
J Neurochem. 1998;
71:741
-748
[138] HOOGENRAAD TU. Dithiocarbamates and Parkinson’s disease.
Lancet. 1988;
1:767
[139] HOROWITZ MP, GREENAMYRE JT. Gene-environment interactions in Parkinson’s disease: the importance of animal modeling.
Clin Pharmacol Ther. 2010;
88:467
-474
[140] HUFF RA, CORCORAN JJ, ANDERSON JK, ABOU-DONIA MB. Chlorpyrifos oxon binds directly to muscarinic receptors and inhibits cAMP accumulation in rat striatum.
J Pharmacol Exp Ther. 1994;
269:329
-335
[141] IANNONE M, CIRIOLO MR, ROTILIO G, NISTICO G. Intra-nigral infusion of Cu-free superoxide dismutase prevents paraquat-induced behavioural stimulation and ECoG epileptogenic discharges in rats.
Neuropharmacology. 1991;
30:893
-898
[142] ICENOGLE LM, CHRISTOPHER NC, BLACKWELDER WP, CALDWELL DP, QIAO D, et coll. Behavioral alterations in adolescent and adult rats caused by a brief subtoxic exposure to chlorpyrifos during neurulation.
Neurotoxicol Teratol. 2004;
26:95
-101
[143] IKEDA T, NAGATA K, SHONO T, NARAHASHI T. Dieldrin and picrotoxinin modulation of GABA(A) receptor single channels.
Neuroreport. 1998;
9:3189
-3195
[144] JOHNSON DE, SEIDLER FJ, SLOTKIN TA. Early biochemical detection of delayed neurotoxicity resulting from developmental exposure to chloropyrifos.
Brain Res Bull. 1998;
45:143
-147
[145] JOHNSON MK, GLYNN P. Neuropathy target esterase (NTE) and organophosphorus-induced delayed polyneuropathy (OPIDP): recent advances.
Toxicol Lett. 1995;
82-83:459
-463
[146] KAKKO I, TOIMELA T, TAHTI H. The toxicity of pyrethroid compounds in neural cell cultures studied with total ATP, mitochondrial enzyme activity and microscopic photographing.
Environ Toxicol Pharmacol. 2004;
15:95
-102
[147] KANG MJ, GIL SJ, KOH HC. Paraquat induces alternation of the dopamine catabolic pathways and glutathione levels in the substantia nigra of mice.
Toxicol Lett. 2009;
188:148
-152
[148] KANTHASAMY AG, KITAZAWA M, KANTHASAMY A, ANANTHARAM V. Dieldrin-induced neurotoxicity: relevance to Parkinson’s disease pathogenesis.
Neurotoxicology. 2005;
26:701
-719
[149] KAREN DJ, LI W, HARP PR, GILLETTE JS, BLOOMQUIST JR. Striatal dopaminergic pathways as a target for the insecticides permethrin and chlorpyrifos.
Neurotoxicology. 2001;
22:811
-817
[150] KIM KY, KIM DS, LEE SK, LEE IK, KANG JH, et coll. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans.
Environ Health Perspect. 2010;
118:370
-374
[151] KIM RH, SMITH PD, ALEYASIN H, HAYLEY S, MOUNT MP, et coll. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress.
Proc Natl Acad Sci USA. 2005;
102:5215
-5220
[152] KIM S, HWANG J, LEE WH, HWANG DY, SUK K. Role of protein kinase Cdelta in paraquat-induced glial cell death.
J Neurosci Res. 2008;
86:2062
-2070
[153] KING TD, JOPE RS. Inhibition of glycogen synthase kinase-3 protects cells from intrinsic but not extrinsic oxidative stress.
Neuroreport. 2005;
16:597
-601
[154] KIRBY ML, BARLOW RL, BLOOMQUIST JR. Selective effects of cyclodiene insecticides on dopamine release in mammalian synaptosomes.
Toxicol Appl Pharmacol. 2002;
181:89
-92
[155] KITAZAWA M, ANANTHARAM V, KANTHASAMY A, KANTHASAMY AG. Dieldrin promotes proteolytic cleavage of poly(ADP-ribose) polymerase and apoptosis in dopaminergic cells: protective effect of mitochondrial anti-apoptotic protein Bcl-2.
Neurotoxicology. 2004;
25:589
-598
[156] KITAZAWA M, ANANTHARAM V, KANTHASAMY AG, 2003 . Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration.
Neuroscience. 119;
945:964
-
[157] KITAZAWA M, ANANTHARAM V, KANTHASAMY AG. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells.
Free Radic Biol Med. 2001;
31:1473
-1485
[158] KLINTWORTH H, NEWHOUSE K, LI T, CHOI WS, FAIGLE R, et coll. Activation of c-Jun N-terminal protein kinase is a common mechanism underlying paraquat- and rotenone-induced dopaminergic cell apoptosis.
Toxicol Sci. 2007;
97:149
-162
[159] KORDOWER JH, KANAAN NM, CHU Y, SURESH BABU R, STANSELL J 3RD, et coll. Failure of proteasome inhibitor administration to provide a model of Parkinson’s disease in rats and monkeys.
Ann Neurol. 2006;
60:264
-268
[160] KROEMER G, BLOMGREN K. Mitochondrial cell death control in familial Parkinson disease.
PLoS Biol. 2007;
5:e206
[161] LAFUENTE A, CABALEIRO T, CARIDE A, ESQUIFINO AI. Toxic effects of methoxychlor administered subcutaneously on the hypothalamic-pituitary-testicular axis in adult rats.
Food Chem Toxicol. 2008;
46:1570
-1575
[162] LANGSTON JW, BALLARD P, TETRUD JW, IRWIN I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis.
Science. 1983;
219:979
-980
[163] LAUDER JM, SCHAMBRA UB. Morphogenetic roles of acetylcholine.
Environ Health Perspect. 1999;
107 (suppl 1):65
-69
[164] LAVARA-CULEBRAS E, PARICIO N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits.
Gene. 2007;
400:158
-165
[165] LAZARINI CA, FLORIO JC, LEMONICA IP, BERNARDI MM. Effects of prenatal exposure to deltamethrin on forced swimming behavior, motor activity, and striatal dopamine levels in male and female rats.
Neurotoxicol Teratol. 2001;
23:665
-673
[166] LECOUTEUR DG, MCLEAN AJ, TAYLOR MC, WOODHAM BL, BOARD PG. Pesticides and Parkinson’s disease.
Biomed Pharmacother. 1999;
53:122
-130
[167] LEE CG, TANG K, CHEUNG YB, WONG LP, TAN C et coll. MDR1, the blood-brain barrier transporter, is associated with Parkinson’s disease in ethnic Chinese.
J Med Genet. 2004;
41:e60
[168] LEE JH, LEE DH, LEE HS, CHOI JS, KIM KW, et coll. Deguelin inhibits human hepatocellular carcinoma by antiangiogenesis and apoptosis.
Oncol Rep. 2008;
20:129
-134
[169] LEVIN ED, ADDY N, NAKAJIMA A, CHRISTOPHER NC, SEIDLER FJ, et coll. Persistent behavioral consequences of neonatal chlorpyrifos exposure in rats.
Brain Res Dev Brain Res. 2001;
130:83
-89
[170] LI J, SPLETTER ML, JOHNSON DA, WRIGHT LS, SVENDSEN CN, et coll. Rotenone-induced caspase 9/3-independent and -dependent cell death in undifferentiated and differentiated human neural stem cells.
J Neurochem. 2005;
92:462
-476
[171] LI WF, COSTA LG, RICHTER RJ, HAGEN T, SHIH DM, et coll. Catalytic efficiency determines the in-vivo efficacy of PON1 for detoxifying organophosphorus compounds.
Pharmacogenetics. 2000;
10:767
-779
[172] LI X, SUN AY. Paraquat induced activation of transcription factor AP-1 and apoptosis in PC12 cells.
J Neural Transm. 1999;
106:1
-21
[173] LIM ML, MERCER LD, NAGLEY P, BEART PM. Rotenone and MPP+ preferentially redistribute apoptosis-inducing factor in apoptotic dopamine neurons.
Neuroreport . 2007;
18:307
-312
[174] LING Z, CHANG QA, TONG CW, LEURGANS SE, LIPTON JW, et coll. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally.
Exp Neurol. 2004;
190:373
-383
[175] LIU Y, ZHAO H, LI H, KALYANARAMAN B, NICOLOSI AC, et coll. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries.
Circ Res. 2003;
93:573
-580
[176] LOTTI M, MORETTO A. Organophosphate-induced delayed polyneuropathy.
Toxicol Rev. 2005;
24:37
-49
[177] LOTTI M. The pathogenesis of organophosphate polyneuropathy.
Crit Rev Toxicol. 1991;
21:465
-487
[178] LUNDVIG D, LINDERSSON E, JENSEN PH. Pathogenic effects of alpha-synuclein aggregation.
Brain Res Mol Brain Res. 2005;
134:3
-17
[179] MALAVIYA M, HUSAIN R, SETH PK. Perinatal effects of two pyrethroid insecticides on brain neurotransmitter function in the neonatal rat.
Vet Hum Toxicol. 1993;
35:119
-122
[180] MANNING-BOG AB, MCCORMACK AL, LI J, UVERSKY VN, FINK AL, et coll. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein.
J Biol Chem. 2002;
277:1641
-1644
[181] MANNING-BOG AB, MCCORMACK AL, PURISAI MG, BOLIN LM, DI MONTE DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration.
J Neurosci. 2003;
23:3095
-3099
[182] MARINOVICH M, GUIZZETTI M, GHILARDI F, VIVIANI B, CORSINI E, et coll. Thyroid peroxidase as toxicity target for dithiocarbamates.
Arch Toxicol. 1997;
71:508
-512
[183] MCCORMACK AL, ATIENZA JG, LANGSTON JW, DI MONTE DA. Decreased susceptibility to oxidative stress underlies the resistance of specific dopaminergic cell populations to paraquat-induced degeneration.
Neuroscience. 2006;
141:929
-937
[184] MCCORMACK AL, DI MONTE DA. Effects of L-dopa and other amino acids against paraquat-induced nigrostriatal degeneration.
J Neurochem. 2003;
85:82
-86
[185] MCCORMACK AL, THIRUCHELVAM M, MANNING-BOG AB, THIFFAULT C, LANGSTON JW, et coll. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat.
Neurobiol Dis. 2002;
10:119
-127
[186] MCGREW DM, IRWIN I, LANGSTON JW. Ethylenebisdithiocarbamate enhances MPTP-induced striatal dopamine depletion in mice.
Neurotoxicology. 2000;
21:309
-312
[187] MCNAUGHT KS, PERL DP, BROWNELL AL, OLANOW CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease.
Ann Neurol. 2004;
56:149
-162
[188] MCNAUGHT KS, SHASHIDHARAN P, PERL DP, JENNER P, OLANOW CW. Aggresome-related biogenesis of Lewy bodies.
Eur J Neurosci. 2002;
16:2136
-2148
[189] MECO G, BONIFATI V, VANACORE N, FABRIZIO E. Parkinsonism after chronic exposure to the fungicide maneb (manganese ethylene-bis-dithiocarbamate).
Scand J Work Environ Health. 1994;
20:301
-305
[190] MEISLER MH, KEARNEY J, OTTMAN R, ESCAYG A. Identification of epilepsy genes in human and mouse.
Annu Rev Genet. 2001;
35:567
-588
[191] MENZIES FM, YENISETTI SC, MIN KT. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress.
Curr Biol. 2005;
15:1578
-1582
[192] MEULENER M, WHITWORTH AJ, ARMSTRONG-GOLD CE, RIZZU P, HEUTINK P, et coll. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease.
Curr Biol. 2005;
15:1572
-1577
[193] MILLER DW, HAGUE SM, CLARIMON J, BAPTISTA M, GWINN-HARDY K, et coll. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication.
Neurology. 2004;
62:1835
-1838
[194] MILLER GW, ERICKSON JD, PEREZ JT, PENLAND SN, MASH DC, et coll. Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson’s disease.
Exp Neurol. 1999a;
156:138
-148
[195] MILLER GW, KIRBY ML, LEVEY AI, BLOOMQUIST JR. Heptachlor alters expression and function of dopamine transporters.
Neurotoxicology. 1999b;
20:631
-637
[196] MILLER RL, SUN GY, SUN AY. Cytotoxicity of paraquat in microglial cells: Involvement of PKCdelta- and ERK1/2-dependent NADPH oxidase.
Brain Res. 2007;
1167:129
-139
[197] MINELLI A, CONTE C, GROTTELLI S, BELLEZZA I, EMILIANI C, et coll. Cyclo(His-Pro) up-regulates heme oxygenase 1 via activation of Nrf2-ARE signalling.
J Neurochem. 2009;
111:956
-966
[198] MOHAMMAD FK, ST OMER VE. Developing rat brain monoamine levels following in utero exposure to a mixture of 2,4-dichlorophenoxyacetic and 2,4,5-trichlorophenoxyacetic acids.
Toxicol Lett . 1985;
29:215
-223
[199] MONIZ AC, BERNARDI MM, SOUZA-SPINOSA HS, PALERMO-NETO J. Effects of exposure to a pyrethroid insecticide during lactation on the behavior of infant and adult rats.
Braz J Med Biol Res. 1990;
23:45
-48
[200] MONNET-TSCHUDI F, ZURICH MG, SCHILTER B, COSTA LG, HONEGGER P. Maturation-dependent effects of chlorpyrifos and parathion and their oxygen analogs on acetylcholinesterase and neuronal and glial markers in aggregating brain cell cultures.
Toxicol Appl Pharmacol. 2000;
165:175
-183
[201] MOREIRA EG, YU X, ROBINSON JF, GRIFFITH W, HONG SW, et coll. Toxicogenomic profiling in maternal and fetal rodent brains following gestational exposure to chlorpyrifos.
Toxicol Appl Pharmacol. 2010;
245:310
-325
[202] MORENO M, CANADAS F, CARDONA D, SUNOL C, CAMPA L, et coll. Long-term monoamine changes in the striatum and nucleus accumbens after acute chlorpyrifos exposure.
Toxicol Lett. 2008;
176:162
-167
[203] MORETTO A, COLOSIO C. Biochemical and toxicological evidence of neurological effects of pesticides: the example of Parkinson’s disease.
Neurotoxicology. 2011;
32:383
-391
[204] MOSER VC. Dose-response and time-course of neurobehavioral changes following oral chlorpyrifos in rats of different ages.
Neurotoxicol Teratol. 2000;
22:713
-723
[205] MURRAY RE, GIBSON JE. Paraquat disposition in rats, guinea pigs and monkeys.
Toxicol Appl Pharmacol . 1974;
27:283
-291
[206] MUTCH E, DALY AK, WILLIAMS FM. The Relationship between PON1 phenotype and PON1-192 genotype in detoxification of three oxons by human liver.
Drug Metab Dispos. 2007;
35:315
-320
[207] NARENDRA D, KANE LA, HAUSER DN, FEARNLEY IM, YOULE RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both.
Autophagy. 2010a;
6:1090
-1106
[208] NARENDRA D, TANAKA A, SUEN DF, YOULE RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy.
J Cell Biol. 2008;
183:795
-803
[209] NARENDRA DP, JIN SM, TANAKA A, SUEN DF, GAUTIER CA, et coll. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin.
PLoS Biol. 2010b;
8:e1000298
[210] NARENDRA DP, YOULE RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control.
Antioxid Redox Signal. 2011;
14:1929
-1938
[211] NASUTI C, GABBIANELLI R, FALCIONI ML, DI STEFANO A, SOZIO P, et coll. Dopaminergic system modulation, behavioral changes, and oxidative stress after neonatal administration of pyrethroids.
Toxicology. 2007;
229:194
-205
[212] NAYLOR JL, WIDDOWSON PS, SIMPSON MG, FARNWORTH M, ELLIS MK, et coll. Further evidence that the blood/brain barrier impedes paraquat entry into the brain.
Hum Exp Toxicol. 1995;
14:587
-594
[213] NEWHOUSE K, HSUAN SL, CHANG SH, CAI B, WANG Y, et coll. Rotenone-induced apoptosis is mediated by p38 and JNK MAP kinases in human dopaminergic SH-SY5Y cells.
Toxicol Sci. 2004;
79:137
-146
[214] NICKLAS WJ, SAPORITO M, BASMA A, GELLER HM, HEIKKILA RE. Mitochondrial mechanisms of neurotoxicity.
Ann N Y Acad Sci. 1992;
648:28
-36
[215] NISO-SANTANO M, GONZALEZ-POLO RA, BRAVO-SAN PEDRO JM, GOMEZ-SANCHEZ R, LASTRES-BECKER I, et coll. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: modulation by the Nrf2/Trx axis.
Free Radic Biol Med. 2010;
48:1370
-1381
[216] NISO-SANTANO M, MORAN JM, GARCIA-RUBIO L, GOMEZ-MARTIN A, GONZALEZ-POLO RA, et coll. Low concentrations of paraquat induces early activation of extracellular signal-regulated kinase 1/2, protein kinase B, and c-Jun N-terminal kinase 1/2 pathways: role of c-Jun N-terminal kinase in paraquat-induced cell death.
Toxicol Sci. 2006;
92:507
-515
[217] NORRIS EH, URYU K, LEIGHT S, GIASSON BI, TROJANOWSKI JQ, et coll. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model.
Am J Pathol. 2007;
170:658
-666
[218] O’LEARY KA, EDWARDS RJ, TOWN MM, BOOBIS AR. Genetic and other sources of variation in the activity of serum paraoxonase/diazoxonase in humans: consequences for risk from exposure to diazinon.
Pharmacogenet Genomics. 2005;
15:51
-60
[219] OLIVIER K, JR., LIU J, POPE C. Inhibition of forskolin-stimulated cAMP formation in vitro by paraoxon and chlorpyrifos oxon in cortical slices from neonatal, juvenile, and adult rats.
J Biochem Mol Toxicol . 2001;
15:263
-269
[220] OSAKADA F, HASHINO A, KUME T, KATSUKI H, KANEKO S, et coll. Alpha-tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons.
Neuropharmacology. 2004;
47:904
-915
[221] OSAKADA F, HASHINO A, KUME T, KATSUKI H, KANEKO S, et coll. Neuroprotective effects of alpha-tocopherol on oxidative stress in rat striatal cultures.
Eur J Pharmacol. 2003;
465:15
-22
[222] OSSOWSKA K, WARDAS J, KUTER K, NOWAK P, DABROWSKA J, et coll. Influence of paraquat on dopaminergic transporter in the rat brain.
Pharmacol Rep. 2005a;
57:330
-335
[223] OSSOWSKA K, WARDAS J, SMIALOWSKA M, KUTER K, LENDA T, et coll. A slowly developing dysfunction of dopaminergic nigrostriatal neurons induced by long-term paraquat administration in rats: an animal model of preclinical stages of Parkinson’s disease?.
Eur J Neurosci. 2005b;
22:1294
-1304
[224] PALACINO JJ, SAGI D, GOLDBERG MS, KRAUSS S, MOTZ C, et coll. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice.
J Biol Chem. 2004;
279:18614
-18622
[225] PEI W, LIOU AK, CHEN J. Two caspase-mediated apoptotic pathways induced by rotenone toxicity in cortical neuronal cells.
FASEB J. 2003;
17:520
-522
[226] PENG J, MAO XO, STEVENSON FF, HSU M, ANDERSEN JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway.
J Biol Chem. 2004;
279:32626
-32632
[227] PITTMAN JT, DODD CA, KLEIN BG. Immunohistochemical changes in the mouse striatum induced by the pyrethroid insecticide permethrin.
Int J Toxicol. 2003;
22:359
-370
[228] POMES A, RODRIGUEZ-FARRE E, SUNOL C. Disruption of GABA-dependent chloride flux by cyclodienes and hexachlorocyclohexanes in primary cultures of cortical neurons.
J Pharmacol Exp Ther. 1994;
271:1616
-1623
[229] POPE CN, CHAKRABORTI TK, CHAPMAN ML, FARRAR JD. Long-term neurochemical and behavioral effects induced by acute chlorpyrifos treatment.
Pharmacol Biochem Behav. 1992;
42:251
-256
[230] POVEY AC, JURY F, DIPPNALL WM, SMITH AE, THOMSON S, et coll. GST CYP and PON1 polymorphisms in farmers attributing ill health to organophosphate-containing sheep dip.
Biomarkers. 2007;
12:188
-202
[231] POVEY AC. Gene-environmental interactions and organophosphate toxicity.
Toxicology. 2010;
278:294
-304
[232] PRASAD K, TARASEWICZ E, MATHEW J, STRICKLAND PA, BUCKLEY B, et coll. Toxicokinetics and toxicodynamics of paraquat accumulation in mouse brain.
Exp Neurol. 2009;
215:358
-367
[233] PRASAD K, WINNIK B, THIRUCHELVAM MJ, BUCKLEY B, MIROCHNITCHENKO O, et coll. Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain.
Environ Health Perspect. 2007;
115:1448
-1453
[234] PURISAI MG, MCCORMACK AL, CUMINE S, LI J, ISLA MZ, et coll. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration.
Neurobiol Dis. 2007;
25:392
-400
[235] PURKERSON-PARKER S, MCDANIEL KL, MOSER VC. Dopamine transporter binding in the rat striatum is increased by gestational, perinatal, and adolescent exposure to heptachlor.
Toxicol Sci. 2001;
64:216
-223
[236] QIAO D, SEIDLER FJ, ABREU-VILLACA Y, TATE CA, COUSINS MM, et coll. Chlorpyrifos exposure during neurulation: cholinergic synaptic dysfunction and cellular alterations in brain regions at adolescence and adulthood.
Brain Res Dev Brain Res. 2004;
148:43
-52
[237] QIAO D, SEIDLER FJ, PADILLA S, SLOTKIN TA. Developmental neurotoxicity of chlorpyrifos: what is the vulnerable period?.
Environ Health Perspect. 2002;
110:1097
-1103
[238] QIAO D, SEIDLER FJ, SLOTKIN TA. Developmental neurotoxicity of chlorpyrifos modeled in vitro: comparative effects of metabolites and other cholinesterase inhibitors on DNA synthesis in PC12 and C6 cells.
Environ Health Perspect. 2001;
109:909
-913
[239] QIAO D, SEIDLER FJ, TATE CA, COUSINS MM, SLOTKIN TA. Fetal chlorpyrifos exposure: adverse effects on brain cell development and cholinergic biomarkers emerge postnatally and continue into adolescence and adulthood.
Environ Health Perspect. 2003b;
111:536
-544
[240] QIAO D, SEIDLER FJ, VIOLIN JD, SLOTKIN TA. Nicotine is a developmental neurotoxicant and neuroprotectant: stage-selective inhibition of DNA synthesis coincident with shielding from effects of chlorpyrifos.
Brain Res Dev Brain Res. 2003a;
147:183
-190
[241] QUIK M, O’LEARY K, TANNER CM. Nicotine and Parkinson’s disease: implications for therapy.
Mov Disord. 2008;
23:1641
-1652
[242] RADAD K, RAUSCH WD, GILLE G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration.
Neurochem Int. 2006;
49:379
-386
[243] RAMACHANDIRAN S, HANSEN JM, JONES DP, RICHARDSON JR, MILLER GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation.
Toxicol Sci. 2007;
95:163
-171
[244] RAY DE, FRY JR. A reassessment of the neurotoxicity of pyrethroid insecticides.
Pharmacol Ther. 2006;
111:174
-193
[245] REN Y, FENG J. Rotenone selectively kills serotonergic neurons through a microtubule-dependent mechanism.
J Neurochem. 2007;
103:303
-311
[246] RICCERI L, MARKINA N, VALANZANO A, FORTUNA S, COMETA MF, et coll. Developmental exposure to chlorpyrifos alters reactivity to environmental and social cues in adolescent mice.
Toxicol Appl Pharmacol. 2003;
191:189
-201
[247] RICCERI L, VENEROSI A, CAPONE F, COMETA MF, LORENZINI P, et coll. Developmental neurotoxicity of organophosphorous pesticides: fetal and neonatal exposure to chlorpyrifos alters sex-specific behaviors at adulthood in mice.
Toxicol Sci. 2006;
93:105
-113
[248] RICHARDSON JR, CAUDLE WM, WANG M, DEAN ED, PENNELL KD, et coll. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson’s disease.
FASEB J. 2006;
20:1695
-1697
[249] RICHARDSON JR, QUAN Y, SHERER TB, GREENAMYRE JT, MILLER GW. Paraquat neurotoxicity is distinct from that of MPTP and rotenone.
Toxicol Sci. 2005;
88:193
-201
[250] RICHTER RJ, JARVIK GP, FURLONG CE. Paraoxonase 1 (PON1) status and substrate hydrolysis.
Toxicol Appl Pharmacol. 2009;
235:1
-9
[251] RIVERA S, ROSA R, MARTINEZ E, SUNOL C, SERRANO MT, et coll. Behavioral and monoaminergic changes after lindane exposure in developing rats.
Neurotoxicol Teratol. 1998;
20:155
-160
[252] ROJO AI, CAVADA C, DE SAGARRA MR, CUADRADO A. Chronic inhalation of rotenone or paraquat does not induce Parkinson’s disease symptoms in mice or rats.
Exp Neurol. 2007;
208:120
-126
[253] ROSSI L, MARCHESE E, LOMBARDO MF, ROTILIO G, CIRIOLO MR. Increased susceptibility of copper-deficient neuroblastoma cells to oxidative stress-mediated apoptosis.
Free Radic Biol Med. 2001;
30:1177
-1187
[254] ROSSO SB, CACERES AO, DE DUFFARD AM, DUFFARD RO, QUIROGA S. 2,4-Dichlorophenoxyacetic acid disrupts the cytoskeleton and disorganizes the Golgi apparatus of cultured neurons.
Toxicol Sci. 2000;
56:133
-140
[255] ROY TS, ANDREWS JE, SEIDLER FJ, SLOTKIN TA. Chlorpyrifos elicits mitotic abnormalities and apoptosis in neuroepithelium of cultured rat embryos.
Teratology. 1998;
58:62
-68
[256] RUGBJERG K, RITZ B, KORBO L, MARTINUSSEN N, OLSEN JH. Risk of Parkinson’s disease after hospital contact for head injury: population based case-control study.
BMJ. 2008;
337:a2494
[257] RYU EJ, HARDING HP, ANGELASTRO JM, VITOLO OV, RON D, et coll. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease.
J Neurosci. 2002;
22:10690
-10698
[258] SAHA S, GUILLILY MD, FERREE A, LANCETA J, CHAN D, et coll. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans.
J Neurosci. 2009;
29:9210
-9218
[259] SAINT-PIERRE M, TREMBLAY ME, SIK A, GROSS RE, CICCHETTI F. Temporal effects of paraquat/maneb on microglial activation and dopamine neuronal loss in older rats.
J Neurochem. 2006;
98:760
-772
[260] SAMS C, COCKER J, LENNARD MS. Biotransformation of chlorpyrifos and diazinon by human liver microsomes and recombinant human cytochrome P450s (CYP).
Xenobiotica. 2004;
34:861
-873
[261] SANCHEZ-RAMOS J, FACCA A, BASIT A, SONG S. Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures.
Exp Neurol. 1998;
150:263
-271
[262] SANCHEZ-SANTED F, CANADAS F, FLORES P, LOPEZ-GRANCHA M, CARDONA D. Long-term functional neurotoxicity of paraoxon and chlorpyrifos: behavioural and pharmacological evidence.
Neurotoxicol Teratol. 2004;
26:305
-317
[263] SCHMUCK G, ROHRDANZ E, TRAN-THI QH, KAHL R, SCHLUTER G. Oxidative stress in rat cortical neurons and astrocytes induced by paraquat in vitro.
Neurotox Res. 2002;
4:1
-13
[264] SCHUH RA, LEIN PJ, BECKLES RA, JETT DA. Noncholinesterase mechanisms of chlorpyrifos neurotoxicity: altered phosphorylation of Ca2+/cAMP response element binding protein in cultured neurons.
Toxicol Appl Pharmacol. 2002;
182:176
-185
[265] SEATON TA, COOPER JM, SCHAPIRA AH. Free radical scavengers protect dopaminergic cell lines from apoptosis induced by complex I inhibitors.
Brain Res. 1997;
777:110
-118
[266] SHAFER TJ, MEYER DA, CROFTON KM. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs.
Environ Health Perspect. 2005;
113:123
-136
[267] SHAIKH SB, NICHOLSON LF. Effects of chronic low dose rotenone treatment on human microglial cells.
Mol Neurodegener. 2009;
4:55
[268] SHARMA RP, WINN DS, LOW JB. Toxic, neurochemical and behavioral effects of dieldrin exposure in mallard ducks.
Arch Environ Contam Toxicol. 1976;
5:43
-53
[269] SHEETS LP, DOHERTY JD, LAW MW, REITER LW, CROFTON KM. Age-dependent differences in the susceptibility of rats to deltamethrin.
Toxicol Appl Pharmacol. 1994;
126:186
-190
[270] SHEETS LP. A consideration of age-dependent differences in susceptibility to organophosphorus and pyrethroid insecticides.
Neurotoxicology. 2000;
21:57
-63
[271] SHERER TB, BETARBET R, GREENAMYRE JT. Environment, mitochondria, and Parkinson’s disease.
Neuroscientist. 2002;
8:192
-197
[272] SHERER TB, BETARBET R, KIM JH, GREENAMYRE JT. Selective microglial activation in the rat rotenone model of Parkinson’s disease.
Neurosci Lett. 2003a;
341:87
-90
[273] SHERER TB, BETARBET R, TESTA CM, SEO BB, RICHARDSON JR, et coll. Mechanism of toxicity in rotenone models of Parkinson’s disease.
J Neurosci. 2003b;
23:10756
-10764
[274] SHIMADA H, FURUNO H, HIRAI K, KOYAMA J, ARIYAMA J, et coll. Paraquat detoxicative system in the mouse liver postmitochondrial fraction.
Arch Biochem Biophys. 2002;
402:149
-157
[275] SHIMIZU K, MATSUBARA K, OHTAKI K, SHIONO H. Paraquat leads to dopaminergic neural vulnerability in organotypic midbrain culture.
Neurosci Res. 2003;
46:523
-532
[276] SHIMIZU K, OHTAKI K, MATSUBARA K, AOYAMA K, UEZONO T, et coll. Carrier-mediated processes in blood--brain barrier penetration and neural uptake of paraquat.
Brain Res. 2001;
906:135
-142
[277] SHINOMIYA N, SHINOMIYA M. Dichlorodiphenyltrichloroethane suppresses neurite outgrowth and induces apoptosis in PC12 pheochromocytoma cells.
Toxicol Lett. 2003;
137:175
-183
[278] SLOTKIN TA, BODWELL BE, LEVIN ED, SEIDLER FJ. Neonatal exposure to low doses of diazinon: long-term effects on neural cell development and acetylcholine systems.
Environ Health Perspect. 2008a;
116:340
-348
[279] SLOTKIN TA, BODWELL BE, RYDE IT, LEVIN ED, SEIDLER FJ. Exposure of neonatal rats to parathion elicits sex-selective impairment of acetylcholine systems in brain regions during adolescence and adulthood.
Environ Health Perspect. 2008b;
116:1308
-1314
[280] SLOTKIN TA, SEIDLER FJ. Developmental neurotoxicants target neurodifferentiation into the serotonin phenotype: Chlorpyrifos, diazinon, dieldrin and divalent nickel.
Toxicol Appl Pharmacol. 2008c;
233:211
-219
[281] SLOTKIN TA, SEIDLER FJ, FUMAGALLI F. Exposure to organophosphates reduces the expression of neurotrophic factors in neonatal rat brain regions: similarities and differences in the effects of chlorpyrifos and diazinon on the fibroblast growth factor superfamily.
Environ Health Perspect. 2007;
115:909
-916
[282] SLOTKIN TA, TATE CA, COUSINS MM, SEIDLER FJ. Functional alterations in CNS catecholamine systems in adolescence and adulthood after neonatal chlorpyrifos exposure.
Brain Res Dev Brain Res. 2002;
133:163
-173
[283] SLOTKIN TA, COUSINS MM, TATE CA, SEIDLER FJ. Persistent cholinergic presynaptic deficits after neonatal chlorpyrifos exposure.
Brain Res. 2001;
902:229
-243
[284] SLOTKIN TA. Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates.
Toxicol Appl Pharmacol. 2004;
198:132
-151
[285] SONG X, SEIDLER FJ, SALEH JL, ZHANG J, PADILLA S, et coll. Cellular mechanisms for developmental toxicity of chlorpyrifos: targeting the adenylyl cyclase signaling cascade.
Toxicol Appl Pharmacol. 1997;
145:158
-174
[286] SOOK HAN M, SHIN KJ, KIM YH, KIM SH, LEE T, et coll. Thiram and ziram stimulate non-selective cation channel and induce apoptosis in PC12 cells.
Neurotoxicology. 2003;
24:425
-434
[287] STEDEFORD T, CARDOZO-PELAEZ F, NEMETH N, SONG S, HARBISON RD, et coll. Comparison of base-excision repair capacity in proliferating and differentiated PC 12 cells following acute challenge with dieldrin.
Free Radic Biol Med. 2001;
31:1272
-1278
[288] SUGENO N, TAKEDA A, HASEGAWA T, KOBAYASHI M, KIKUCHI A, et coll. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death.
J Biol Chem. 2008;
283:23179
-23188
[289] TAI KK, MCCROSSAN ZA, ABBOTT GW. Activation of mitochondrial ATP-sensitive potassium channels increases cell viability against rotenone-induced cell death.
J Neurochem. 2003;
84:1193
-1200
[290] TAI KK, TRUONG DD. Activation of adenosine triphosphate-sensitive potassium channels confers protection against rotenone-induced cell death: therapeutic implications for Parkinson’s disease.
J Neurosci Res. 2002;
69:559
-566
[291] TAKAHASHI RN, ROGERIO R, ZANIN M. Maneb enhances MPTP neurotoxicity in mice.
Res Commun Chem Pathol Pharmacol. 1989;
66:167
-170
[292] TALTS U, FREDRIKSSON A, ERIKSSON P. Changes in behavior and muscarinic receptor density after neonatal and adult exposure to bioallethrin.
Neurobiol Aging. 1998a;
19:545
-552
[293] TALTS U, TALTS JF, ERIKSSON P. Differential expression of muscarinic subtype mRNAs after exposure to neurotoxic pesticides.
Neurobiol Aging. 1998b;
19:553
-559
[294] TAWARA T, FUKUSHIMA T, HOJO N, ISOBE A, SHIWAKU K, et coll. Effects of paraquat on mitochondrial electron transport system and catecholamine contents in rat brain.
Arch Toxicol. 1996;
70:585
-589
[295] THIFFAULT C, LANGSTON JW, DI MONTE DA. Increased striatal dopamine turnover following acute administration of rotenone to mice.
Brain Res. 2000;
885:283
-288
[296] THIRUCHELVAM M, BROCKEL BJ, RICHFIELD EK, BAGGS RB, CORY-SLECHTA DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson’s disease?.
Brain Res. 2000a;
873:225
-234
[297] THIRUCHELVAM M, MCCORMACK A, RICHFIELD EK, BAGGS RB, TANK AW, et coll. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson’s disease phenotype.
Eur J Neurosci. 2003;
18:589
-600
[298] THIRUCHELVAM M, PROKOPENKO O, CORY-SLECHTA DA, BUCKLEY B, MIROCHNITCHENKO O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype.
J Biol Chem. 2005;
280:22530
-22539
[299] THIRUCHELVAM M, RICHFIELD EK, BAGGS RB, TANK AW, CORY-SLECHTA DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson’s disease.
J Neurosci. 2000b;
20:9207
-9214
[300] THIRUCHELVAM M, RICHFIELD EK, GOODMAN BM, BAGGS RB, CORY-SLECHTA DA. Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype.
Neurotoxicology. 2002;
23:621
-633
[301] THIRUCHELVAM MJ, POWERS JM, CORY-SLECHTA DA, RICHFIELD EK. Risk factors for dopaminergic neuron loss in human alpha-synuclein transgenic mice.
Eur J Neurosci. 2004;
19:845
-854
[302] TORRES-ALTORO MI, MATHUR BN, DRERUP JM, THOMAS R, LOVINGER DM, et coll. Organophosphates dysregulate dopamine signaling, glutamatergic neurotransmission, and induce neuronal injury markers in striatum.
J Neurochem. 2011;
119:303
-313
[303] TRAN V, HOFFMAN N, MOFUNANAYA A, PRYOR SC, OJUGBELE O, et coll. Bifenthrin inhibits neurite outgrowth in differentiating PC12 cells.
Med Sci Monit. 2006;
12:BR57
-BR62
[304] UVERSKY VN, ELIEZER D. Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein.
Curr Protein Pept Sci. 2009;
10:483
-499
[305] UVERSKY VN, LI J, BOWER K, FINK AL. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson’s disease.
Neurotoxicology. 2002;
23:527
-536
[306] UVERSKY VN, LI J, FINK AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease.
FEBS Lett. 2001;
500:105
-108
[307] UVERSKY VN. Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration.
Cell Tissue Res. 2004;
318:225
-241
[308] VACCARI A, SABA P, MOCCI I, RUIU S. Dithiocarbamate pesticides affect glutamate transport in brain synaptic vesicles.
J Pharmacol Exp Ther. 1999;
288:1
-5
[309] VALE C, FONFRIA E, BUJONS J, MESSEGUER A, RODRIGUEZ-FARRE E, et coll. The organochlorine pesticides gamma-hexachlorocyclohexane (lindane), alpha-endosulfan and dieldrin differentially interact with GABA(A) and glycine-gated chloride channels in primary cultures of cerebellar granule cells.
Neuroscience. 2003;
117:397
-403
[310] VED R, SAHA S, WESTLUND B, PERIER C, BURNAM L, et coll. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans.
J Biol Chem. 2005;
280:42655
-42668
[311] VIDAIR CA. Age dependence of organophosphate and carbamate neurotoxicity in the postnatal rat: extrapolation to the human.
Toxicol Appl Pharmacol. 2004;
196:287
-302
[312] VOGT M, BAUER MK, FERRARI D, SCHULZE-OSTHOFF K. Oxidative stress and hypoxia/reoxygenation trigger CD95 (APO-1/Fas) ligand expression in microglial cells.
FEBS Lett. 1998;
429:67
-72
[313] WAGNER SR, GREENE FE. Dieldrin-induced alterations in biogenic amine content of rat brain.
Toxicol Appl Pharmacol. 1978;
43:45
-55
[314] WANG C, KO HS, THOMAS B, TSANG F, CHEW KC, et coll. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function.
Hum Mol Genet. 2005;
14:3885
-3897
[315] WANG C, LU R, OUYANG X, HO MW, CHIA W, et coll. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities.
J Neurosci. 2007;
27:8563
-8570
[316] WANG L, YANG HJ, XIA YY, FENG ZW. Insulin-like growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3beta/JNK signaling.
Apoptosis. 2010;
15:1470
-1479
[317] WARD TR, MUNDY WR. Organophosphorus compounds preferentially affect second messenger systems coupled to M2/M4 receptors in rat frontal cortex.
Brain Res Bull. 1996;
39:49
-55
[318] WATABE M, NAKAKI T. Mitochondrial complex I inhibitor rotenone inhibits and redistributes vesicular monoamine transporter 2 via nitration in human dopaminergic SH-SY5Y cells.
Mol Pharmacol. 2008;
74:933
-940
[319] WATABE M, NAKAKI T. Rotenone induces apoptosis via activation of bad in human dopaminergic SH-SY5Y cells.
J Pharmacol Exp Ther. 2004;
311:948
-953
[320] WEISSKOPF MG, O’REILLY E, CHEN H, SCHWARZSCHILD MA, ASCHERIO A. Plasma urate and risk of Parkinson’s disease.
Am J Epidemiol. 2007;
166:561
-567
[321] WESTERLUND M, BELIN AC, ANVRET A, HÅKANSSON A, NISSBRANDT H, et coll. Association of a polymorphism in the ABCB1 gene with Parkinson’s disease.
Parkinsonism Relat Disord. 2009;
15:422
-424
[322] WHITNEY KD, SEIDLER FJ, SLOTKIN TA. Developmental neurotoxicity of chlorpyrifos: cellular mechanisms.
Toxicol Appl Pharmacol. 1995;
134:53
-62
[323] WIDDOWSON PS, FARNWORTH MJ, SIMPSON MG, LOCK EA. Influence of age on the passage of paraquat through the blood-brain barrier in rats: a distribution and pathological examination.
Hum Exp Toxicol. 1996a;
15:231
-236
[324] WIDDOWSON PS, FARNWORTH MJ, UPTON R, SIMPSON MG. No changes in behaviour, nigro-striatal system neurochemistry or neuronal cell death following toxic multiple oral paraquat administration to rats.
Hum Exp Toxicol. 1996b;
15:583
-591
[325] WOOD-KACZMAR A, GANDHI S, WOOD NW. Understanding the molecular causes of Parkinson’s disease.
Trends Mol Med. 2006;
12:521
-528
[326] WU A, LIU Y. Prolonged expression of c-Fos and c-Jun in the cerebral cortex of rats after deltamethrin treatment.
Brain Res Mol Brain Res. 2003;
110:147
-151
[327] WU XF, BLOCK ML, ZHANG W, QIN L, WILSON B, et coll. The role of microglia in paraquat-induced dopaminergic neurotoxicity.
Antioxid Redox Signal. 2005;
7:654
-661
[328] YANG W, TIFFANY-CASTIGLIONI E, KOH HC, SON IH. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells.
Toxicol Lett. 2009;
191:203
-210
[329] YANG W, TIFFANY-CASTIGLIONI E. Paraquat-induced apoptosis in human neuroblastoma SH-SY5Y cells: involvement of p53 and mitochondria.
J Toxicol Environ Health. A 2008;
71:289
-299
[330] YANG W, TIFFANY-CASTIGLIONI E. The bipyridyl herbicide paraquat produces oxidative stress-mediated toxicity in human neuroblastoma SH-SY5Y cells: relevance to the dopaminergic pathogenesis.
J Toxicol Environ Health A. 2005;
68:1939
-1961
[331] ZAIDI A, FERNANDES D, BEAN JL, MICHAELIS ML. Effects of paraquat-induced oxidative stress on the neuronal plasma membrane Ca(2+)-ATPase.
Free Radic Biol Med. 2009;
47:1507
-1514
[332] ZEEVALK GD, BERNARD LP, GUILFORD FT. Liposomal-glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells.
Neurochem Res. 2010;
35:1575
-1587
[333] ZHANG H, LIU J, POPE CN. Age-related effects of chlorpyrifos on muscarinic receptor-mediated signaling in rat cortex.
Arch Toxicol. 2002;
75:676
-684
[334] ZHANG J, FITSANAKIS VA, GU G, JING D, AO M, et coll. Manganese ethylene-bis-dithiocarbamate and selective dopaminergic neurodegeneration in rat: a link through mitochondrial dysfunction.
J Neurochem. 2003;
84:336
-346
[335] ZHAO X, SALGADO VL, YEH JZ, NARAHASHI T. Differential actions of fipronil and dieldrin insecticides on GABA-gated chloride channels in cockroach neurons.
J Pharmacol Exp Ther. 2003;
306:914
-924
[336] ZHOU Y, SHIE FS, PICCARDO P, MONTINE TJ, ZHANG J. Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: relevance to Parkinson’s disease.
Neuroscience. 2004;
128:281
-291
[337] ZSCHIEDRICH K, KÖNIG IR, BRÜGGEMANN N, KOCK N, KASTEN M, et coll. MDR1 variants and risk of Parkinson disease. Association with pesticide exposure?.
J Neurol. 2009;
256:115
-120
→ Aller vers SYNTHESE