III. Toxicologie
2013
| ANALYSE |
24-
Mécanismes d’action des pesticides dans les pathologies métaboliques
Le syndrome métabolique regroupe un certain nombre de manifestations pathologiques dont l’obésité, l’intolérance au glucose, une dyslipidémie et une hypertension. Les individus développant ces symptômes présentent un risque accru (multiplié par 5) de développement de diabète de type 2, mais aussi de certains cancers ou de maladies cardiovasculaires. Actuellement, aux États-Unis, le nombre estimé de personnes affectées par cette pathologie est de l’ordre de 27 %. Or, deux tiers des américains sont considérés comme étant en surpoids (1/3 obèses) (Flegal et coll., 2012 ). Ainsi, les extrapolations estiment que plus de 350 millions de personnes pourraient présenter un diabète de type 2 d’ici 2030 (incluant celles développant un diabète sans syndrome métabolique).
). Ainsi, les extrapolations estiment que plus de 350 millions de personnes pourraient présenter un diabète de type 2 d’ici 2030 (incluant celles développant un diabète sans syndrome métabolique).
). Ainsi, les extrapolations estiment que plus de 350 millions de personnes pourraient présenter un diabète de type 2 d’ici 2030 (incluant celles développant un diabète sans syndrome métabolique).Le diabète de type 2 se caractérise par une glycémie élevée, due à une résistance périphérique à l’action de l’insuline, hormone hypoglycémiante. Une conséquence de cette résistance est une hyper-insulinémie (compensant la perte de sensibilité). L’obésité est un facteur majeur contribuant à la mise en place de cette résistance ; celle-ci est probablement due à la présence en quantité anormalement élevée de molécules de signalisation dans le sang, liée à l’hyper-croissance du tissu adipeux. Trois théories non exclusives ont été formulées pour expliquer ce lien : le développement d’un état inflammatoire du tissu adipeux (les cytokines pro-inflammatoires favorisant la résistance vis-à-vis de l’insuline), la fonction endocrine accrue du tissu adipeux (tissu adipeux, tissu de stockage mais également organe endocrine produisant des cytokines, les adipokines jouant un rôle régulateur dans le métabolisme énergétique, la sensibilité à l’insuline comme la résistine) et la capacité de croissance limitée des adipocytes (en partie régulée par certaines cytokines pro-inflammatoires). Par ailleurs, l’obésité s’accompagne d’une diminution de la production d’adiponectine (adipokine dont le taux élevé diminuerait le risque de diabète) (Antuna-Puente et coll., 2008).
).Si l’obésité représente une composante du syndrome métabolique, facteur de risque du diabète de type 2, certains individus dont l’indice de masse corporel est normal, sont susceptibles de développer cette pathologie et à l’inverse, certains individus obèses sont « métaboliquement » sains. D’autres facteurs sont donc susceptibles d’intervenir. C’est probablement le cas de polluants environnementaux dont certains pesticides. Dans cet ouvrage, la communication de Jérémie Botton « Pesticides et pathologies métaboliques : données épidémiologiques » fait le point sur les travaux qui suggèrent des liens potentiels entre l’exposition à certains pesticides (comme à d’autres polluants environnementaux) et différentes manifestations pathologiques du syndrome métabolique. Citons par exemple :
• métabolisme et fonction thyroïdienne : taux sériques de pesticides organochlorés (OC) et hypothyroïdie ou niveau des hormones thyroïdiennes (T4), polymorphismes de la paraoxonase 1 (PON1, impliquée dans le métabolisme des organophosphorés) et dysfonctionnement thyroïdien ;
• obésité et croissance pondérale : taux sériques des OC et obésité, concentrations d’hexachlorobenzène (HCB) ou de dichlorodiphényltrichloroéthane (DDE) dans le sang maternel/cordon ombilical et croissance pondérale chez l’enfant quelques années après la naissance ;
• syndrome métabolique et diabète de type 2 : exposition aux chlorophénoxyherbicides et mortalité par infarctus et/ou diabète de type 2, concentration de certains OC, organophosphorés (dichlorvos, trichlorfon) et herbicides (alachlore, cyanazine) et développement d’un diabète de type 2, concentration de certains pyréthrinoïdes et dérégulation de la glycémie.
Ces études rappellent certaines observations effectuées sur d’autres contaminants environnementaux (dioxines, bisphénol A) (Henriksen et coll., 1997 ; Cranmer et coll., 2000 ; Kern et coll., 2004 ; Fujiyoshi et coll., 2006). L’ensemble de ces données soulève la question des mécanismes potentiels mis en jeu dans l’évolution vers ces pathologies insidieuses.
; Cranmer et coll., 2000 ; Kern et coll., 2004 ; Fujiyoshi et coll., 2006). L’ensemble de ces données soulève la question des mécanismes potentiels mis en jeu dans l’évolution vers ces pathologies insidieuses.Mécanismes potentiels mis en jeu par deux types de polluants dans le développement de pathologies métaboliques
Sur le plan expérimental, peu de données sur pesticides et syndrome métabolique ou pesticides et diabète de type 2 sont disponibles. Des travaux récents menés avec d’autres types de polluants comme la tétrachlorodibenzodioxine (TCDD) ou le bisphénol A (BPA), qui sont susceptibles d’agir via les mêmes voies de signalisation (notamment celles impliquant certains récepteurs nucléaires comme ceux des œstrogènes et des androgènes) que certains pesticides comme les OC – dont les mécanismes sont moins décrits dans la littérature – peuvent permettre de formaliser quelques hypothèses mécanistiques.
Exemple du bisphénol A et des dioxines
Plusieurs études expérimentales suggèrent que le Bisphénol A (BPA) et la TCDD favorisent la mise en place d’une résistance à l’insuline et perturbent ainsi l’homéostasie glucidique (Enan et coll., 1992 ; Ishida et coll., 2005 ; Novelli et coll., 2005 ; Alonso-Magdalena et coll., 2006 et 2010a et b ; Kurita et coll., 2009 ; Ruzzin et coll., 2010). Une exposition au BPA pendant la gestation semble influencer le poids à la naissance des souriceaux et plus tardivement, leur métabolisme glucidique (Rubin et coll., 2009). Ces résultats sont retrouvés chez le rat (Miyawaki et coll., 2007 ; Somm et coll., 2009). Chez des souris adultes exposées à des doses relativement faibles (par exemple : 10 µg/kg, très inférieure à la dose quotidienne acceptée par les agences européennes de 50 µg/kg/jour chez l’Homme), le BPA provoque une hyper-insulinémie (accompagnée simultanément d’une hypoglycémie). Il semble également favoriser une résistance vis-à-vis de l’insuline au niveau périphérique (foie, cellules musculaires et tissu adipeux).
; Ishida et coll., 2005 ; Novelli et coll., 2005 ; Alonso-Magdalena et coll., 2006 et 2010a et b ; Kurita et coll., 2009 ; Ruzzin et coll., 2010). Une exposition au BPA pendant la gestation semble influencer le poids à la naissance des souriceaux et plus tardivement, leur métabolisme glucidique (Rubin et coll., 2009). Ces résultats sont retrouvés chez le rat (Miyawaki et coll., 2007 ; Somm et coll., 2009). Chez des souris adultes exposées à des doses relativement faibles (par exemple : 10 µg/kg, très inférieure à la dose quotidienne acceptée par les agences européennes de 50 µg/kg/jour chez l’Homme), le BPA provoque une hyper-insulinémie (accompagnée simultanément d’une hypoglycémie). Il semble également favoriser une résistance vis-à-vis de l’insuline au niveau périphérique (foie, cellules musculaires et tissu adipeux).Au niveau pancréatique, l’un des mécanismes d’action évoqués pour expliquer l’hyperinsulinémie est celui d’un effet œstrogénique non-génomique (Ropero et coll., 2008). Les doses de BPA utilisées pour cette étude sont faibles et pour certaines inférieures à la dose admissible journalière pour l’Homme (Adachi et coll., 2005 ; Alonso-Magdalena et coll., 2008). Bien que cela soit plus controversé, la TCDD semble elle aussi perturber la sécrétion d’insuline dans différents types de modèles (Weber et coll., 1991a et b ; Enan et coll., 1992 ; Ishida et coll., 2005 ; Kurita et coll., 2009).
). Les doses de BPA utilisées pour cette étude sont faibles et pour certaines inférieures à la dose admissible journalière pour l’Homme (Adachi et coll., 2005 ; Alonso-Magdalena et coll., 2008). Bien que cela soit plus controversé, la TCDD semble elle aussi perturber la sécrétion d’insuline dans différents types de modèles (Weber et coll., 1991a et b ; Enan et coll., 1992 ; Ishida et coll., 2005 ; Kurita et coll., 2009).Sur le phénomène de résistance périphérique, des études expérimentales ont montré qu’une dose de 100 µg/kg de BPA administrée pendant 4 jours à des souris provoquait une intolérance au glucose (et une résistance à l’insuline). Le BPA à très faibles doses (pico/nanomolaires) provoque une réduction de la sécrétion par le tissu adipeux, d’adiponectine qui est considérée comme une molécule anti-inflammatoire augmentant la sensibilité des adipocytes vis-à-vis de l’insuline (Whitehead et coll., 2006 ; Hugo et coll., 2008). À des doses plus élevées, la réduction de la sécrétion de cette hormone a été retrouvée ainsi que d’autres altérations de la fonction adipocytaire ou hépatique (accumulation de lipides hépatiques, différenciation de fibroblastes en adipocytes, baisse de la synthèse de leptine) (Masuno et coll., 2005 ; Wada et coll., 2007 ; Phrakonkham et coll., 2008 ; Kidani el coll., 2010).
; Hugo et coll., 2008). À des doses plus élevées, la réduction de la sécrétion de cette hormone a été retrouvée ainsi que d’autres altérations de la fonction adipocytaire ou hépatique (accumulation de lipides hépatiques, différenciation de fibroblastes en adipocytes, baisse de la synthèse de leptine) (Masuno et coll., 2005 ; Wada et coll., 2007 ; Phrakonkham et coll., 2008 ; Kidani el coll., 2010).Rôle de la signalisation œstrogénique
Le BPA est un des perturbateurs endocriniens qui a été le plus étudié ; certains pesticides OC semblent partager un certain nombre de mécanismes d’action avec celui-ci. L’un des points les plus intéressants soulevés par les études expérimentales utilisant le BPA, est le probable rôle des récepteurs aux œstrogènes (ER ou estrogen receptor en anglais) dans les phénomènes observés. Chez l’Homme, le récepteur existe sous deux formes, alpha et bêta.
Sur le plan physiologique, les effets des œstrogènes sont complexes car ces hormones régulent les fonctions de nombreux tissus (Inserm, 2011). Sur le plan métabolique, les ER participent au développement du cerveau et contribuent ensuite au contrôle de la prise alimentaire. Au niveau périphérique, les ER sont exprimés dans les adipocytes et pré-adipocytes ; leur activation par les œstrogènes conduit notamment à une augmentation du nombre de cellules adipeuses au cours du développement. Paradoxalement, chez les femmes ménopausées (ou chez des rongeurs ovariectomisés), la diminution de la production d’œstrogènes est plutôt associée à une augmentation du volume du tissu adipeux blanc. Le rôle de ces hormones et de leurs récepteurs est avéré par la supplémentation œstrogénique ou la démonstration d’une résistance à l’insuline chez les souris chez lesquelles le gène ER alpha a été invalidé (knock-out). Il est intéressant de noter qu’en fin de grossesse, la femme enceinte développe une résistance à l’insuline (permettant un apport de nutriments au fœtus en quantité suffisante). En parallèle, son pancréas s’adapte à ce nouvel état physiologique en sécrétant une plus grande quantité d’insuline. La signalisation œstrogénique est particulièrement importante pour cette double adaptation. Il est donc important de noter qu’en fonction de l’âge (ou du stade de développement) et du contexte physiologique, les œstrogènes et leurs récepteurs (notamment la forme alpha) ne régulent pas de la même façon, l’homéostasie énergétique de l’organisme.
). Sur le plan métabolique, les ER participent au développement du cerveau et contribuent ensuite au contrôle de la prise alimentaire. Au niveau périphérique, les ER sont exprimés dans les adipocytes et pré-adipocytes ; leur activation par les œstrogènes conduit notamment à une augmentation du nombre de cellules adipeuses au cours du développement. Paradoxalement, chez les femmes ménopausées (ou chez des rongeurs ovariectomisés), la diminution de la production d’œstrogènes est plutôt associée à une augmentation du volume du tissu adipeux blanc. Le rôle de ces hormones et de leurs récepteurs est avéré par la supplémentation œstrogénique ou la démonstration d’une résistance à l’insuline chez les souris chez lesquelles le gène ER alpha a été invalidé (knock-out). Il est intéressant de noter qu’en fin de grossesse, la femme enceinte développe une résistance à l’insuline (permettant un apport de nutriments au fœtus en quantité suffisante). En parallèle, son pancréas s’adapte à ce nouvel état physiologique en sécrétant une plus grande quantité d’insuline. La signalisation œstrogénique est particulièrement importante pour cette double adaptation. Il est donc important de noter qu’en fonction de l’âge (ou du stade de développement) et du contexte physiologique, les œstrogènes et leurs récepteurs (notamment la forme alpha) ne régulent pas de la même façon, l’homéostasie énergétique de l’organisme.Sur le plan mécanistique, dans la cellule bêta-pancréatique, le BPA semble stimuler le récepteur alpha aux œstrogènes, ce qui conduit à une augmentation de l’influx calcique (ou augmentation de la concentration intracellulaire en Ca2+) qui a, pour double conséquence, de stimuler l’activité du facteur de transcription CREB (cAMP Responsive Element Binding protein) et la sécrétion d’insuline (des études expérimentales montrent également que le BPA augmente l’expression de l’insuline sur le long terme) (Alonso-Magdalena et coll., 2008). L’influence du récepteur bêta est encore mal définie dans ce cadre. À l’inverse de la cellule bêta-pancréatique, l’influx calcique semble diminué en présence de BPA dans la cellule alpha, avec pour conséquence, une moindre sécrétion de glucagon (Quesada et coll., 2002 ; Alonso-Magdalena et coll., 2005 et 2006). La voie de signalisation impliquée dans cet influx d’ion calcium serait non-génomique. Ce mode d’activation paraît également important en périphérie dans l’adipocyte où la production de résistine (suspectée de favoriser la résistance à l’insuline) serait stimulée par l’ER alpha et la voie des ERK (Extracellular Regulated Kinases) (Lee et coll., 2008). Ces voies de stimulation non génomiques pourraient impliquer la protéine membranaire GPR30 (un récepteur membranaire des œstrogènes) dont la délétion chez la souris conduit à une intolérance vis-à-vis du glucose et une diminution de la sécrétion d’insuline par le pancréas, stimulée par les œstrogènes (Martensson et coll., 2009) (figure 24.1).
). L’influence du récepteur bêta est encore mal définie dans ce cadre. À l’inverse de la cellule bêta-pancréatique, l’influx calcique semble diminué en présence de BPA dans la cellule alpha, avec pour conséquence, une moindre sécrétion de glucagon (Quesada et coll., 2002 ; Alonso-Magdalena et coll., 2005 et 2006). La voie de signalisation impliquée dans cet influx d’ion calcium serait non-génomique. Ce mode d’activation paraît également important en périphérie dans l’adipocyte où la production de résistine (suspectée de favoriser la résistance à l’insuline) serait stimulée par l’ER alpha et la voie des ERK (Extracellular Regulated Kinases) (Lee et coll., 2008). Ces voies de stimulation non génomiques pourraient impliquer la protéine membranaire GPR30 (un récepteur membranaire des œstrogènes) dont la délétion chez la souris conduit à une intolérance vis-à-vis du glucose et une diminution de la sécrétion d’insuline par le pancréas, stimulée par les œstrogènes (Martensson et coll., 2009) (figure 24.1).
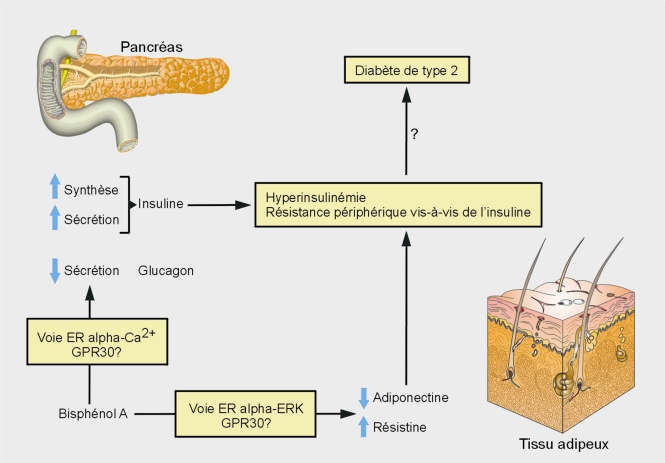
Un autre polluant suspecté de favoriser ces phénomènes est la dioxine de Sévéso ou TCDD. Celle-ci active un récepteur intracellulaire appelé AhR (Aryl hydrocarbon Receptor) qui agit principalement comme un facteur transcriptionnel. Cette protéine peut également se comporter comme un co-activateur du récepteur alpha des œstrogènes quand celui-ci n’est pas lié à un composé agoniste. Ainsi, des ligands du AhR tels que la TCDD pourraient activer la voie de signalisation du ER alpha en l’absence de stimulation œstrogénique. Il faut toutefois noter, qu’en présence d’un agoniste lié au ER, la TCDD agirait plutôt comme une molécule anti-œstrogénique favorisant la dégradation de ce récepteur. Les paradoxes observés dans le cadre des études sur la TCDD pourraient donc s’expliquer par cet effet opposé en fonction de l’état de liaison du ER alpha.
Influence des pesticides organochlorés sur le métabolisme
Dans différents travaux, plusieurs pesticides organochlorés (OC) sont suspectés d’influencer le poids des enfants à la naissance, de favoriser la mise en place d’une obésité, d’un syndrome métabolique voire d’un diabète (type 2) chez l’Homme. Ces effets sont retrouvés chez le singe. Il faut souligner le peu de données chez le rongeur.
Plusieurs OC (dont le chlordane et le DDT, dichlorodiphényltrichloroéthane) ne sont plus utilisés depuis longtemps dans de nombreux pays mais leurs métabolites comme l’oxychlordane ou le DDE, persistent dans l’environnement et s’accumulent dans la chaîne alimentaire. À titre d’exemple, le DDE s’accumule fortement dans le tissu adipeux (coefficient de partition tissu adipeux/sang de 400/1, Muhlebach et coll., 1991). Il en résulte que certaines concentrations seraient supérieures à 10 µM chez l’Homme dans certaines cellules notamment adipocytaires (supérieures, en comparaison, aux concentrations sanguines) (Mullerova et Kopecky, 2007). Les études s’intéressant aux processus de différenciation adipogénique et menées à faibles et fortes doses de polluants sont donc toutes deux, pertinentes.
). Il en résulte que certaines concentrations seraient supérieures à 10 µM chez l’Homme dans certaines cellules notamment adipocytaires (supérieures, en comparaison, aux concentrations sanguines) (Mullerova et Kopecky, 2007). Les études s’intéressant aux processus de différenciation adipogénique et menées à faibles et fortes doses de polluants sont donc toutes deux, pertinentes.Effets sur la différenciation adipocytaire
Plusieurs études menées in vitro suggèrent que la différenciation adipocytaire et sa production de cytokines/adipokines sont perturbées par des doses assez élevées d’OC (DDT, aldrine, endrine, dieldrine) (Moreno-Aliaga et Matsumura, 1999 et 2002 ; Howell et coll., 2011). Ainsi, Howell et coll. (2011), en utilisant la lignée murine 3T3-L1 (modèle de différenciation adipocytaire passant du stade pré-adipocyte au stade adipocyte), montrent que la dieldrine altère la différenciation à forte dose (20 µM mais pas à 2 µM), ce qui n’est pas le cas des fortes doses de DDE ou d’oxychlordane.
et 2002 ; Howell et coll., 2011). Ainsi, Howell et coll. (2011), en utilisant la lignée murine 3T3-L1 (modèle de différenciation adipocytaire passant du stade pré-adipocyte au stade adipocyte), montrent que la dieldrine altère la différenciation à forte dose (20 µM mais pas à 2 µM), ce qui n’est pas le cas des fortes doses de DDE ou d’oxychlordane.La capture basale d’acides gras libres (non stimulée par l’insuline) par ces mêmes cellules différenciées, est altérée par l’oxychlordane (20 µM), le DDE (2 µM) ou la dieldrine (2-20 µM). De même, la secrétion des adipokines, adiponectine, résistine et leptine est augmentée dans les cellules 3T3-L1 différenciées et les adipocytes exposées au DDE.
L’interprétation de l’ensemble de ces résultats est délicate mais suggère de manière forte une perturbation de la différenciation adipocytaire pour plusieurs OC à forte dose (Howell et coll., 2011). Ces résultats sont retrouvés par Moreno-Aliaga et coll., qui montrent qu’un autre OC, l’endrine, inhibe la différenciation adipocytaire (3T3-L1) à des doses là aussi assez élevées (10-30 µM). L’effet de l’aldrine et de la dieldrine est moins important et uniquement observé à forte concentration.
). Ces résultats sont retrouvés par Moreno-Aliaga et coll., qui montrent qu’un autre OC, l’endrine, inhibe la différenciation adipocytaire (3T3-L1) à des doses là aussi assez élevées (10-30 µM). L’effet de l’aldrine et de la dieldrine est moins important et uniquement observé à forte concentration.Les auteurs proposent un mécanisme d’action pour l’endrine passant par une déplétion des niveaux intracellulaires des récepteurs C/EBP alpha et de PPAR gamma qui sont des facteurs majeurs de différenciation de cette lignée (Moreno-Aliaga et Matsumura, 1999). Les effets observés par cette étude ne semblent toutefois pas être généralisables à tous les OC. Ainsi, les mêmes auteurs montrent que le DDT favorise la différenciation de cellules 3T3-L1. Cet effet passe par l’augmentation des niveaux nucléaires de récepteurs PPAR gamma et C/EBP alpha et de ce fait, par la liaison augmentée de ce dernier à ces éléments de réponse (Moreno-Aliaga et Matsumura, 2002).
). Les effets observés par cette étude ne semblent toutefois pas être généralisables à tous les OC. Ainsi, les mêmes auteurs montrent que le DDT favorise la différenciation de cellules 3T3-L1. Cet effet passe par l’augmentation des niveaux nucléaires de récepteurs PPAR gamma et C/EBP alpha et de ce fait, par la liaison augmentée de ce dernier à ces éléments de réponse (Moreno-Aliaga et Matsumura, 2002).En résumé, les OC semblent perturber la différenciation adipocytaire à forte dose. Cependant, les observations réalisées avec un OC ne peuvent être généralisées à l’ensemble des membres de cette famille chimique. Par ailleurs, le choix du type ou de la lignée cellulaire pour ces études semble lui aussi crucial. Ainsi, de fortes doses de DDT (20 µM) favorisent la différenciation de cellules 3T3-L1 et inhibent partiellement le programme de différenciation (Moreno-Aliaga et Matsumura, 2002) d’une autre lignée de pré-adipocytes (3T3-F442A). Ce paradoxe peut s’expliquer par des différences significatives entre ces deux lignées au cours du processus de différenciation (inducteurs différents, sécrétion de leptine, niveau de récepteurs C/EBPalpha)1
. Des études associant à la fois des modèles animaux et cellulaires semblent donc nécessaires pour mieux comprendre les effets des OC sur le métabolisme énergétique.
) d’une autre lignée de pré-adipocytes (3T3-F442A). Ce paradoxe peut s’expliquer par des différences significatives entre ces deux lignées au cours du processus de différenciation (inducteurs différents, sécrétion de leptine, niveau de récepteurs C/EBPalpha)1
. Des études associant à la fois des modèles animaux et cellulaires semblent donc nécessaires pour mieux comprendre les effets des OC sur le métabolisme énergétique.Effets de faibles doses : interventions des récepteurs stéroïdiens ?
Chez des rats Sprague-Dawley, un régime riche en lipides (supplémentation en huile de saumon contenant plusieurs polluants organiques persistants ou POPs dont des pesticides OC) conduit à une prise de poids plus élevée, une augmentation de certains dépôts graisseux au niveau viscéral et à une modification importante de certains paramètres lipidiques sanguins, contrairement à un régime similaire contenant sensiblement moins de POPs (Ruzzin et coll., 2010). Ces modifications s’accompagnent à terme d’une hyper-insulinémie, d’une résistance à l’insuline périphérique et d’une stéatose hépatique. L’analyse de l’expression de gènes candidats dans les foies de ces animaux révèle que les POPs activent la lipogenèse et l’inflammation et réduisent la fonction oxydative mitochondriale. Les auteurs ont ensuite utilisé individuellement les POPs présents dans l’huile de saumon contaminée et démontré notamment que les OC (et dans une moindre mesure, certains PCBs) à des doses très faibles (1-10-100 nM) étaient capables de bloquer l’action de l’insuline sur des adipocytes en culture2
.
). Ces modifications s’accompagnent à terme d’une hyper-insulinémie, d’une résistance à l’insuline périphérique et d’une stéatose hépatique. L’analyse de l’expression de gènes candidats dans les foies de ces animaux révèle que les POPs activent la lipogenèse et l’inflammation et réduisent la fonction oxydative mitochondriale. Les auteurs ont ensuite utilisé individuellement les POPs présents dans l’huile de saumon contaminée et démontré notamment que les OC (et dans une moindre mesure, certains PCBs) à des doses très faibles (1-10-100 nM) étaient capables de bloquer l’action de l’insuline sur des adipocytes en culture2
.En résumé, les effets physiopathologiques à faible dose rappellent ceux évoqués plus haut pour le BPA et laissent supposer une possibilité d’intervention des récepteurs stéroïdiens (œstrogènes en particulier) dans ces phénomènes.
Activation du récepteur PXR
Les OC ont également été décrits comme des activateurs du récepteur de xénobiotiques PXR (Coumoul et coll., 2002). Bien que les conséquences de cette activation n’aient pas été étudiées d’un point de vue métabolique, des études ont été menées sur ce récepteur à l’aide d’autres ligands : ainsi, la rifampicine (qui active le PXR humain mais pas le PXR de rongeurs) favorise la stéatose hépatique chez des patients auxquels il est prescrit comme antibiotique anti-tuberculeux (Morere et coll., 1975). Chez les rongeurs (rat et souris), l’activation du PXR (ou de CAR, un autre récepteur de xénobiotiques) a également un effet pro-stéatosique au niveau hépatique (Nakamura et coll., 2007 ; Zhou et coll., 2008). Le PXR régule par ailleurs l’expression d’enzymes du métabolisme des xénobiotiques dont des cytochromes P450 (exemple : CYP3A4 chez l’Homme) qui peuvent catalyser la dégradation de certaines hormones stéroïdes (dont les œstrogènes). Ceci pourrait avoir des conséquences importantes sur l’adiposité (voir l’effet d’une déplétion œstrogénique, par exemple chez la femme ménopausée) (Kretschmer et coll., 2005). L’activation du PXR (ou de CAR) pourrait toutefois avoir un effet paradoxal car leur stimulation semble inhiber l’expression hépatique d’enzymes de la néoglucogenèse limitant ainsi une production excessive de glucose (Argaud et coll., 1991 ; Kodama et coll., 2004 ; Zhou et coll., 2008). Cette régulation passerait par des interactions avec d’autres facteurs transcriptionnels (forkhead box O1 ou FoxO1, forkhead box A2 ou FoxA2, cAMP-response element binding protein ou C/EBP) ou co-activateurs (peroxisome proliferator activated receptor gamma coactivator-1 alpha ou PGC-1 alpha) et conduirait au niveau hépatique à la diminution d’activité de la néoglucogenèse mais aussi à celle de la ß-oxydation des acides gras (associée à une lipogenèse plus active) expliquant ainsi la stéatose hépatique (Konno et coll., 2008).
). Bien que les conséquences de cette activation n’aient pas été étudiées d’un point de vue métabolique, des études ont été menées sur ce récepteur à l’aide d’autres ligands : ainsi, la rifampicine (qui active le PXR humain mais pas le PXR de rongeurs) favorise la stéatose hépatique chez des patients auxquels il est prescrit comme antibiotique anti-tuberculeux (Morere et coll., 1975). Chez les rongeurs (rat et souris), l’activation du PXR (ou de CAR, un autre récepteur de xénobiotiques) a également un effet pro-stéatosique au niveau hépatique (Nakamura et coll., 2007 ; Zhou et coll., 2008). Le PXR régule par ailleurs l’expression d’enzymes du métabolisme des xénobiotiques dont des cytochromes P450 (exemple : CYP3A4 chez l’Homme) qui peuvent catalyser la dégradation de certaines hormones stéroïdes (dont les œstrogènes). Ceci pourrait avoir des conséquences importantes sur l’adiposité (voir l’effet d’une déplétion œstrogénique, par exemple chez la femme ménopausée) (Kretschmer et coll., 2005). L’activation du PXR (ou de CAR) pourrait toutefois avoir un effet paradoxal car leur stimulation semble inhiber l’expression hépatique d’enzymes de la néoglucogenèse limitant ainsi une production excessive de glucose (Argaud et coll., 1991 ; Kodama et coll., 2004 ; Zhou et coll., 2008). Cette régulation passerait par des interactions avec d’autres facteurs transcriptionnels (forkhead box O1 ou FoxO1, forkhead box A2 ou FoxA2, cAMP-response element binding protein ou C/EBP) ou co-activateurs (peroxisome proliferator activated receptor gamma coactivator-1 alpha ou PGC-1 alpha) et conduirait au niveau hépatique à la diminution d’activité de la néoglucogenèse mais aussi à celle de la ß-oxydation des acides gras (associée à une lipogenèse plus active) expliquant ainsi la stéatose hépatique (Konno et coll., 2008).Cet effet paradoxal rappelle certaines observations réalisées chez des patients obèses contraints à une perte de poids et dont les concentrations plasmatiques de POPs (en partie relargués par le tissu adipeux du fait de sa diminution de volume) ont été mesurées pendant la période d’amaigrissement. Chez ces individus, une corrélation positive est observée entre la quantité de POP plasmatique libérée par le tissu adipeux et l’amélioration de sensibilité à l’insuline. Le mécanisme expliquant cet effet pourrait aussi être une inhibition de la néoglucogenèse (Kim et coll., 2010).
).En résumé, les récepteurs de xénobiotiques, PXR et CAR, apparaissent comme modulateurs dans des processus parfois paradoxaux de régulation métabolique mais dont les conséquences sont concordantes sur le plan clinique.
En conclusion, les données épidémiologiques suggèrent que l’exposition à certains pesticides conduit à l’apparition de pathologies métaboliques dont le diabète de type 2. Bien que parcellaires et parfois contradictoires (en cas de généralisation abusive ; voir l’effet divergent du DDT sur les deux lignées cellulaires 3T3), les données expérimentales permettent de proposer un schéma rendant compte des effets potentiels de certains pesticides organochlorés sur le développement de ces maladies (figure 24.2).
).Plusieurs voies de signalisation pourraient ainsi être impliquées parmi lesquelles celles impliquant les récepteurs aux œstrogènes. Au niveau pancréatique, les régulations non génomiques semblent jouer un rôle essentiel notamment dans la sécrétion de glucagon et d’insuline. Cependant, selon les modèles et protocoles utilisés, l’effet des pesticides organochlorés sur les voies de signalisation des récepteurs nucléaires peut varier. Ainsi, dans les études in vivo, les niveaux sériques d’œstrogènes varient grandement en fonction de l’âge, du stade de développement, du type d’animaux utilisés (cycles ovariens très différents). Par ailleurs, les doses utilisées peuvent conduire à des effets très différents et pour un composé non pesticide comme le bisphénol A, une action a parfois été relevée à faibles ou fortes doses mais pas à des concentrations intermédiaires (relation en U). Dans le cadre des pesticides organochlorés, un autre niveau de complexité est celui de la persistance qui conduit à une accumulation de certains d’entre eux et de leurs métabolites (DDT et DDE par exemple) dans le tissu adipeux rendant ainsi potentiellement pertinentes certaines études à forte dose réalisées sur des modèles de ce tissu.
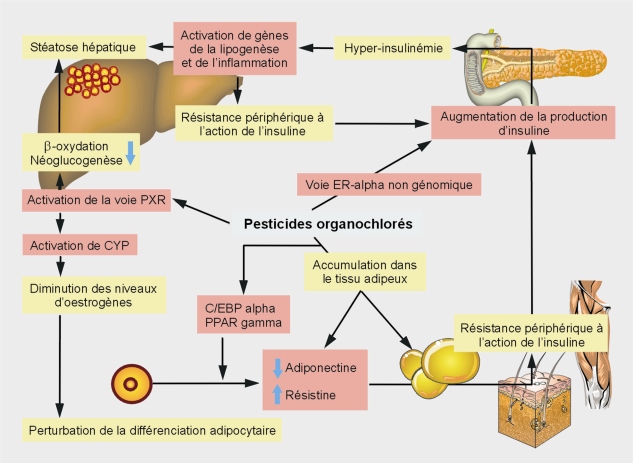
En plus des récepteurs aux œstrogènes, de nombreuses autres cibles des pesticides existent et ont été décrites comme modulatrices de fonctions métaboliques. C’est le cas des récepteurs de xénobiotiques, AhR, PXR et CAR (qui semblent être impliqués dans des processus certes paradoxaux de régulation métabolique mais dont les conséquences sont concordantes sur le plan clinique) mais également d’autres récepteurs de molécules endogènes comme le GR (glucocorticoid receptor) et les PPAR (peroxisome proliferator-activated receptor). Plus de 200 pesticides ont, ainsi, été testés quant à leur potentiel d’activation des gènes cibles des PPAR alpha ou gamma. Si aucun n’active le PPAR gamma, le diclofop-méthyl (un herbicide, inhibiteur de l’acétyl-coA carboxylase) et deux pyréthrines augmentent l’expression de gènes cibles du récepteur alpha à des niveaux équivalents à ceux d’agonistes classiques (Takeuchi et coll., 2006). Ce type de protocole mérite probablement d’être reproduit en s’intéressant à d’autres récepteurs et complété par des approches à la fois in vitro et in vivo (modèles cellulaires de différenciation adipocytaire, suivi de l’évolution de la masse adipeuse chez l’animal par des techniques non invasives comme l’imagerie par IRM) afin de progresser dans la connaissance des mécanismes mis en jeu dans les associations observées entre pesticides et pathologies métaboliques.
). Ce type de protocole mérite probablement d’être reproduit en s’intéressant à d’autres récepteurs et complété par des approches à la fois in vitro et in vivo (modèles cellulaires de différenciation adipocytaire, suivi de l’évolution de la masse adipeuse chez l’animal par des techniques non invasives comme l’imagerie par IRM) afin de progresser dans la connaissance des mécanismes mis en jeu dans les associations observées entre pesticides et pathologies métaboliques.Bibliographie
[1] ADACHI T, YASUDA K, MORI C, YOSHINAGA M, AOKI N, et coll. Promoting insulin secretion in pancreatic islets by means of bisphenol A and nonylphenol via intracellular estrogen receptors.
Food Chem Toxicol. 2005;
43:713- 719
[2] ALONSO-MAGDALENA P, LARIBI O, ROPERO AB, FUENTES E, RIPOLL C, et coll. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans.
Environ Health Perspect. 2005;
113:969- 977
[3] ALONSO-MAGDALENA P, MORIMOTO S, RIPOLL C, FUENTES E, NADAL A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance.
Environ Health Perspect. 2006;
114:106- 112
[4] ALONSO-MAGDALENA P, ROPERO AB, CARRERA MP, CEDERROTH CR, BAQUIE M, et coll. Pancreatic insulin content regulation by the estrogen receptor ER alpha.
PLoS One. 2008;
3:e2069
[5] ALONSO-MAGDALENA P, VIEIRA E, SORIANO S, MENES L, BURKS D, et coll. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring.
Environ Health Perspect. 2010a;
118:1243- 1250
[6] ALONSO-MAGDALENA P, ROPERO AB, SORIANO S, QUESADA I, NADAL A. Bisphenol-A: a new diabetogenic factor?.
Hormones (Athens). 2010b;
9:118- 126
[7] ANTUNA-PUENTE B, FEVE B, FELLAHI S, BASTARD JP. Adipokines: the missing link between insulin resistance and obesity.
Diabetes Metab. 2008;
34:2- 11
[8] ARGAUD D, HALIMI S, CATELLONI F, LEVERVE XM. Inhibition of gluconeogenesis in isolated rat hepatocytes after chronic treatment with phenobarbital.
Biochem J. 1991;
280 (Pt 3):663- 669
[9] COUMOUL X, DIRY M, BAROUKI R. PXR-dependent induction of human CYP3A4 gene expression by organochlorine pesticides.
Biochem Pharmacol. 2002;
64:1513- 1519
[10] CRANMER M, LOUIE S, KENNEDY RH, KERN PA, FONSECA VA. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is associated with hyperinsulinemia and insulin resistance.
Toxicol Sci. 2000;
56:431- 436
[11] ENAN E, LIU PC, MATSUMURA F. TCDD (2,3,7,8-tetrachlorodibenzo-P-dioxin) causes reduction in glucose uptake through glucose transporters on the plasma membrane of the guinea pig adipocyte.
J Environ Sci Health B. 1992;
27:495- 510
[12] FLEGAL KM, CARROLL MD, KIT BK, OGDEN CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010.
JAMA. 2012;
307:491- 497
[13] FUJIYOSHI PT, MICHALEK JE, MATSUMURA F. Molecular epidemiologic evidence for diabetogenic effects of dioxin exposure in U.S. Air force veterans of the Vietnam war.
Environ Health Perspect. 2006;
114:1677- 1683
[14] HENRIKSEN GL, KETCHUM NS, MICHALEK JE, SWABY JA. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand.
Epidemiology. 1997;
8:252- 258
[15] HOWELL G, III, MANGUM L. Exposure to bioaccumulative organochlorine compounds alters adipogenesis, fatty acid uptake, and adipokine production in NIH3T3-L1 cells.
Toxicol In. 2011;
25:394- 402
[16] HUGO ER, BRANDEBOURG TD, WOO JG, LOFTUS J, ALEXANDER JW, et coll. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes.
Environ Health Perspect. 2008;
116:1642- 1647
[17]INSERM. Reproduction et environnement. Collection Expertise Collective Inserm, Editions Inserm, Paris.
2011;
713pp.
[18] ISHIDA T, KAN-O S, MUTOH J, TAKEDA S, ISHII Y, et coll. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced change in intestinal function and pathology: evidence for the involvement of arylhydrocarbon receptor-mediated alteration of glucose transportation.
Toxicol Appl Pharmacol. 2005;
205:89- 97
[19] KERN PA, SAID S, JACKSON WG, JR, MICHALEK JE. Insulin sensitivity following agent orange exposure in Vietnam veterans with high blood levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin.
J Clin Endocrinol Metab. 2004;
89:4665- 4672
[20] KIDANI T, KAMEI S, MIYAWAKI J, AIZAWA J, SAKAYAMA K, et coll. Bisphenol A downregulates Akt signaling and inhibits adiponectin production and secretion in 3T3-L1 adipocytes.
J Atheroscler Thromb. 2010;
17:834- 843
[21] KIM MJ, MARCHAND P, HENEGAR C, ANTIGNAC JP, ALILI R et coll. Fate and complex pathogenic effects of dioxins and polychlorinated biphenyls in obese subjects before and after drastic weight loss.
Environ Health Perspect. 2011;
119:377- 383
[22] KODAMA S, KOIKE C, NEGISHI M, YAMAMOTO Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes.
Mol Cell Biol. 2004;
24:7931- 7940
[23] KONNO Y, NEGISHI M, KODAMA S. The roles of nuclear receptors CAR and PXR in hepatic energy metabolism.
Drug Metab Pharmacokinet. 2008;
23:8- 13
[24] KRETSCHMER XC, BALDWIN WS. CAR and PXR: xenosensors of endocrine disrupters?.
Chem Biol Interact. 2005;
155:111- 128
[25] KURITA H, YOSHIOKA W, NISHIMURA N, KUBOTA N, KADOWAKI T, et coll. Aryl hydrocarbon receptor-mediated effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on glucose-stimulated insulin secretion in mice.
J Appl Toxicol. 2009;
29:689- 694
[26] LEE MJ, LIN H, LIU CW, WU MH, LIAO WJ, et coll. Octylphenol stimulated resistin gene expression in 3T3-L1 adipocytes via the estrogen receptor and extracellular signal-regulated kinase pathways.
Am J Physiol Cell Physiol. 2008;
294:C1542- C1551
[27] MARTENSSON UE, SALEHI SA, WINDAHL S, GOMEZ MF, SWARD K, et coll. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice.
Endocrinology. 2009;
150:687- 698
[28] MASUNO H, IWANAMI J, KIDANI T, SAKAYAMA K, HONDA K. Bisphenol a accelerates terminal differentiation of 3T3-L1 cells into adipocytes through the phosphatidylinositol 3-kinase pathway.
Toxicol Sci. 2005;
84:319- 327
[29] MIYAWAKI J, SAKAYAMA K, KATO H, YAMAMOTO H, MASUNO H. Perinatal and postnatal exposure to bisphenol a increases adipose tissue mass and serum cholesterol level in mice.
J Atheroscler Thromb. 2007;
14:245- 252
[30] MORENO-ALIAGA MJ, MATSUMURA F. Effects of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)-ethane (p,p’-DDT) on 3T3-L1 and 3T3-F442A adipocyte differentiation.
Biochemical pharmacology. 2002;
63:997- 1007
[31] MORENO-ALIAGA MJ, MATSUMURA F. Endrin inhibits adipocyte differentiation by selectively altering expression pattern of CCAAT/enhancer binding protein-alpha in 3T3-L1 cells.
Molecular pharmacology. 1999;
56:91- 101
[32] MORERE P, NOUVET G, STAIN JP, PAILLOT B, METAYER J, et coll. Information obtained by liver biopsy in 100 tuberculous patients.
La semaine des hôpitaux : organe fondé par l’Association d’enseignement médical des hôpitaux de Paris. 1975;
51:20952102
[33] MUHLEBACH S, MOOR MJ, WYSS PA, BICKEL MH. Kinetics of distribution and elimination of DDE in rats.
Xenobiotica; the fate of foreign compounds in biological systems. 1991;
21:111- 120
[34] MULLEROVA D, KOPECKY J. White adipose tissue: storage and effector site for environmental pollutants.
Physiological research / Academia Scientiarum Bohemoslovaca. 2007;
56:375- 381
[35] NAKAMURA K, MOORE R, NEGISHI M, SUEYOSHI T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver.
The Journal of biological chemistry. 2007;
282:9768- 9776
[36] NOVELLI M, PIAGGI S, DE TATA V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets.
Toxicol Lett. 2005;
156:307- 314
[37] OGDEN CL, CARROLL MD, CURTIN LR, MCDOWELL MA, TABAK CJ, FLEGAL KM. Prevalence of overweight and obesity in the United States, 1999-2004.
JAMA. 2006;
295:1549- 1555
[38] PHRAKONKHAM P, VIENGCHAREUN S, BELLOIR C, LOMBES M, ARTUR Y, et coll. Dietary xenoestrogens differentially impair 3T3-L1 preadipocyte differentiation and persistently affect leptin synthesis.
The Journal of steroid biochemistry and molecular biology. 2008;
110:95- 103
[39] QUESADA I, FUENTES E, VISO-LEON MC, SORIA B, RIPOLL C, et coll. Low doses of the endocrine disruptor bisphenol-A and the native hormone 17beta-estradiol rapidly activate transcription factor CREB.
FASEB J. 2002;
16:1671- 1673
[40] ROPERO AB, ALONSO-MAGDALENA P, GARCIA-GARCIA E, RIPOLL C, FUENTES E, et coll. Bisphenol-A disruption of the endocrine pancreas and blood glucose homeostasis.
International journal of andrology. 2008;
31:194- 200
[41] RUBIN BS, SOTO AM. Bisphenol A: Perinatal exposure and body weight.
Molecular and cellular endocrinology. 2009;
304:55- 62
[42] RUZZIN J, PETERSEN R, MEUGNIER E, MADSEN L, LOCK EJ, et coll. Persistent organic pollutant exposure leads to insulin resistance syndrome.
Environ Health Perspect. 2010;
118:465- 471
[43] SOMM E, SCHWITZGEBEL VM, TOULOTTE A, CEDERROTH CR, COMBESCURE C, et coll. Perinatal exposure to bisphenol a alters early adipogenesis in the rat.
Environ Health Perspect. 2009;
117:1549- 1555
[44] TAKEUCHI S, MATSUDA T, KOBAYASHI S, TAKAHASHI T, KOJIMA H. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma and quantitative analysis of in vivo induction pathway.
Toxicol Appl Pharmacol. 2006;
217:235- 244
[45] WADA K, SAKAMOTO H, NISHIKAWA K, SAKUMA S, NAKAJIMA A, et coll. Life style-related diseases of the digestive system: endocrine disruptors stimulate lipid accumulation in target cells related to metabolic syndrome.
Journal of pharmacological sciences. 2007;
105:133- 137
[46] WEBER LW, LEBOFSKY M, GREIM H, ROZMAN K. Key enzymes of gluconeogenesis are dose-dependently reduced in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats.
Archives of toxicology. 1991a;
65:119- 123
[47] WEBER LW, LEBOFSKY M, STAHL BU, GORSKI JR, MUZI G, et coll. Reduced activities of key enzymes of gluconeogenesis as possible cause of acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats.
Toxicology. 1991b;
66:133- 144
[48] WHITEHEAD JP, RICHARDS AA, HICKMAN IJ, MACDONALD GA, PRINS JB. Adiponectin--a key adipokine in the metabolic syndrome.
Diabetes, obesity & metabolism. 2006;
8:264- 280
[49] ZHOU J, FEBBRAIO M, WADA T, ZHAI Y, KURUBA R, et coll. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis.
Gastroenterology. 2008;
134:556- 567
→ Aller vers SYNTHESE