I. Substances psychoactives
2014
| ANALYSE |
6-
Vulnérabilité des adolescents aux addictions et corrélats neurobiologiques
Un défi majeur dans la compréhension des troubles liés à la consommation excessive de substances psychoactives est de déterminer pourquoi une certaine proportion d’individus (10-30 %) perd la liberté de s’abstenir alors que la majorité est en capacité de résister à l’addiction. La recherche sur les mécanismes neurobiologiques qui sous-tendent le développement de l’addiction a produit ces dernières années un changement radical de la compréhension de ce trouble « bio-psycho-médico-social ». Les processus neurobiologiques à la base de la vulnérabilité et l’interaction complexe entre les différents facteurs mis en jeu sont maintenant mieux compris. Parmi ceux-ci, on retrouve les facteurs génétiques (environ 50 % du risque de développer une addiction, avec par exemple leur participation dans la sensibilité aux effets plaisants, la tolérance et le métabolisme), les facteurs développementaux (vie intra-utérine, enfance, adolescence) et les facteurs environnementaux (stress, drogue, social, familial, culturel) qui sont impliqués dans le risque de perdre le contrôle de la consommation et de présenter un usage compulsif (Naassila, 2008 ). Les cibles cellulaires et moléculaires des substances psychoactives ainsi que les systèmes de neurotransmission et les circuits cérébraux impliqués dans le développement de l’addiction sont maintenant bien identifiés. Les prises initiales sont associées au plaisir qu’elles produisent (renforcement positif) par l’intermédiaire de l’augmentation de la transmission dopaminergique du circuit mésocorticolimbique. À long terme, la perte de contrôle de la consommation, la consommation pour se soulager des effets néfastes du sevrage (renforcement négatif), la compulsion (prise en dépit des effets néfastes) et le craving qui est un facteur précipitant la rechute, sont associés à d’autres circuits cérébraux. Ces circuits sont impliqués notamment dans les fonctions exécutives (contrôle inhibiteur, prise de décision, attribution de la plus-value), la mémoire (conditionnement, habitude), la récompense, le conditionnement, la motivation (énergie, dynamisme), l’humeur (réactivité au stress, état hédonique) et l’intéroception (conscience des perturbations interne à l’organisme). Le chemin qui mène du plaisir à la dépendance passe très certainement par des phénomènes liés d’une part à l’automatisme dans lequel le comportement initialement motivé devient par la suite une habitude et d’autre part à une augmentation progressive de la motivation à consommer (tolérance inverse ou sensibilisation) (Vanderschuren et Pierce, 2010). L’automatisme ou le comportement lié à l’habitude n’est pas affecté lorsque la récompense (drogue) devient moins attractive (dévaluée) ou si l’action n’est plus nécessaire à l’obtention de la récompense. Enfin, la consommation chronique de drogue entraîne de nombreuses adaptations cérébrales (processus opposants ou allostasie) qui pourraient expliquer la présence d’un affect négatif (dysphorie, anxiété, irritabilité) chez le sujet dépendant (Koob et Le Moal, 2008).
). Les cibles cellulaires et moléculaires des substances psychoactives ainsi que les systèmes de neurotransmission et les circuits cérébraux impliqués dans le développement de l’addiction sont maintenant bien identifiés. Les prises initiales sont associées au plaisir qu’elles produisent (renforcement positif) par l’intermédiaire de l’augmentation de la transmission dopaminergique du circuit mésocorticolimbique. À long terme, la perte de contrôle de la consommation, la consommation pour se soulager des effets néfastes du sevrage (renforcement négatif), la compulsion (prise en dépit des effets néfastes) et le craving qui est un facteur précipitant la rechute, sont associés à d’autres circuits cérébraux. Ces circuits sont impliqués notamment dans les fonctions exécutives (contrôle inhibiteur, prise de décision, attribution de la plus-value), la mémoire (conditionnement, habitude), la récompense, le conditionnement, la motivation (énergie, dynamisme), l’humeur (réactivité au stress, état hédonique) et l’intéroception (conscience des perturbations interne à l’organisme). Le chemin qui mène du plaisir à la dépendance passe très certainement par des phénomènes liés d’une part à l’automatisme dans lequel le comportement initialement motivé devient par la suite une habitude et d’autre part à une augmentation progressive de la motivation à consommer (tolérance inverse ou sensibilisation) (Vanderschuren et Pierce, 2010). L’automatisme ou le comportement lié à l’habitude n’est pas affecté lorsque la récompense (drogue) devient moins attractive (dévaluée) ou si l’action n’est plus nécessaire à l’obtention de la récompense. Enfin, la consommation chronique de drogue entraîne de nombreuses adaptations cérébrales (processus opposants ou allostasie) qui pourraient expliquer la présence d’un affect négatif (dysphorie, anxiété, irritabilité) chez le sujet dépendant (Koob et Le Moal, 2008).
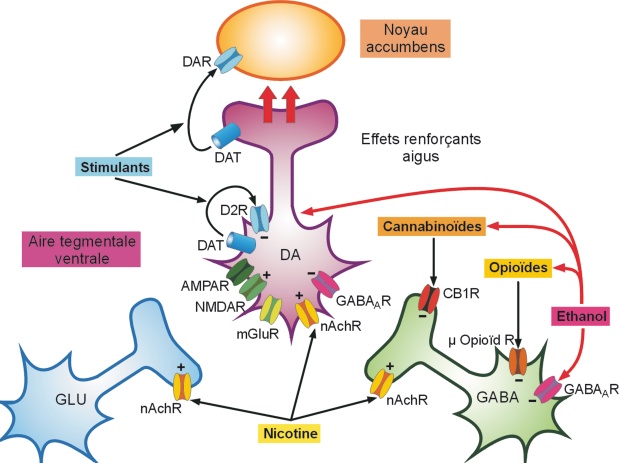
). Les cibles cellulaires et moléculaires des substances psychoactives ainsi que les systèmes de neurotransmission et les circuits cérébraux impliqués dans le développement de l’addiction sont maintenant bien identifiés. Les prises initiales sont associées au plaisir qu’elles produisent (renforcement positif) par l’intermédiaire de l’augmentation de la transmission dopaminergique du circuit mésocorticolimbique. À long terme, la perte de contrôle de la consommation, la consommation pour se soulager des effets néfastes du sevrage (renforcement négatif), la compulsion (prise en dépit des effets néfastes) et le craving qui est un facteur précipitant la rechute, sont associés à d’autres circuits cérébraux. Ces circuits sont impliqués notamment dans les fonctions exécutives (contrôle inhibiteur, prise de décision, attribution de la plus-value), la mémoire (conditionnement, habitude), la récompense, le conditionnement, la motivation (énergie, dynamisme), l’humeur (réactivité au stress, état hédonique) et l’intéroception (conscience des perturbations interne à l’organisme). Le chemin qui mène du plaisir à la dépendance passe très certainement par des phénomènes liés d’une part à l’automatisme dans lequel le comportement initialement motivé devient par la suite une habitude et d’autre part à une augmentation progressive de la motivation à consommer (tolérance inverse ou sensibilisation) (Vanderschuren et Pierce, 2010). L’automatisme ou le comportement lié à l’habitude n’est pas affecté lorsque la récompense (drogue) devient moins attractive (dévaluée) ou si l’action n’est plus nécessaire à l’obtention de la récompense. Enfin, la consommation chronique de drogue entraîne de nombreuses adaptations cérébrales (processus opposants ou allostasie) qui pourraient expliquer la présence d’un affect négatif (dysphorie, anxiété, irritabilité) chez le sujet dépendant (Koob et Le Moal, 2008).Les effets plaisants, dits encore récompensants des drogues sont relayés par la libération de dopamine dans le noyau accumbens (Nac) par les terminaisons synaptiques en provenance des neurones de l’aire tegmentale ventrale (ATV) du circuit mésocorticolimbique (figure 6.1). En fait, les drogues ne font qu’usurper le rôle des comportements (alimentation, sexualité) ou les effets des substances qui produisent naturellement du plaisir via la libération de dopamine. Cet effet aigu des drogues, qui augmente la concentration de dopamine extracellulaire, s’explique par différents mécanismes dont la diminution du tonus inhibiteur qu’exercent les neurones GABAergiques sur les neurones dopaminergiques de l’ATV, la libération d’opioïdes et d’endocannabinoïdes endogènes, et une action directe sur les neurones dopaminergiques en augmentant leur fréquence de décharge. Des études d’imagerie cérébrale chez l’Homme ont établi que les effets plaisants des drogues étaient corrélés à la quantité de dopamine libérée. La dopamine libérée sous-tend non seulement les effets plaisants mais intervient aussi dans des phénomènes beaucoup plus complexes d’attribution de la « plus-value » (« valeur incitatrice ») associée à la drogue. Elle relaye également le mécanisme de prédiction de l’erreur lorsqu’un indice contextuel (neutre) qui a été apparié de manière répétée à la prise de drogue n’est plus suivi par la délivrance du produit (Schultz, 2010). Ainsi, un indice contextuel (« cue ») associé à la prise de drogue, se voit attribuer (après une période de conditionnement) la valeur de la drogue elle-même et est capable de précipiter la rechute en provoquant un envahissement de l’esprit par le désir impérieux, urgent et irrépressible de consommer la drogue. Il faut noter que la libération de dopamine dans la partie dorsale du striatum (région qui semble jouer un rôle majeur dans les aspects liés aux habitudes et aux automatismes caractéristiques du comportement addictif et de la recherche compulsive de drogue) induite par l’indice contextuel pourrait même être supérieure à celle induite par la drogue elle-même. Cela expliquerait pourquoi l’environnement jouerait un rôle si important. Ces indices environnementaux associés au désir de consommer la drogue entraînent des réponses conditionnées en contrôlant la transmission dopaminergique et maintiennent une forte motivation à consommer. Chez un sujet sain, une augmentation de la neurotransmission dopaminergique est observée après la prise aiguë de drogue. Les études d’imagerie cérébrale ont montré, chez les sujets dépendants, des niveaux supra-physiologiques de dopamine dans le Nac associés à une diminution marquée de la fonction dopaminergique, avec notamment une réduction des taux de récepteurs D2 de la dopamine. Le déficit en récepteurs D2 de la dopamine pourrait jouer un rôle majeur dans la vulnérabilité à devenir dépendant. La diminution de la transmission dopaminergique est à l’origine de la baisse généralisée de la sensibilité du système de la récompense aux effets des récompenses naturelles. En revanche, les effets de la drogue et l’apprentissage conditionné entre ces effets et les stimuli neutres associés (indices contextuels) se renforcent.
). En fait, les drogues ne font qu’usurper le rôle des comportements (alimentation, sexualité) ou les effets des substances qui produisent naturellement du plaisir via la libération de dopamine. Cet effet aigu des drogues, qui augmente la concentration de dopamine extracellulaire, s’explique par différents mécanismes dont la diminution du tonus inhibiteur qu’exercent les neurones GABAergiques sur les neurones dopaminergiques de l’ATV, la libération d’opioïdes et d’endocannabinoïdes endogènes, et une action directe sur les neurones dopaminergiques en augmentant leur fréquence de décharge. Des études d’imagerie cérébrale chez l’Homme ont établi que les effets plaisants des drogues étaient corrélés à la quantité de dopamine libérée. La dopamine libérée sous-tend non seulement les effets plaisants mais intervient aussi dans des phénomènes beaucoup plus complexes d’attribution de la « plus-value » (« valeur incitatrice ») associée à la drogue. Elle relaye également le mécanisme de prédiction de l’erreur lorsqu’un indice contextuel (neutre) qui a été apparié de manière répétée à la prise de drogue n’est plus suivi par la délivrance du produit (Schultz, 2010). Ainsi, un indice contextuel (« cue ») associé à la prise de drogue, se voit attribuer (après une période de conditionnement) la valeur de la drogue elle-même et est capable de précipiter la rechute en provoquant un envahissement de l’esprit par le désir impérieux, urgent et irrépressible de consommer la drogue. Il faut noter que la libération de dopamine dans la partie dorsale du striatum (région qui semble jouer un rôle majeur dans les aspects liés aux habitudes et aux automatismes caractéristiques du comportement addictif et de la recherche compulsive de drogue) induite par l’indice contextuel pourrait même être supérieure à celle induite par la drogue elle-même. Cela expliquerait pourquoi l’environnement jouerait un rôle si important. Ces indices environnementaux associés au désir de consommer la drogue entraînent des réponses conditionnées en contrôlant la transmission dopaminergique et maintiennent une forte motivation à consommer. Chez un sujet sain, une augmentation de la neurotransmission dopaminergique est observée après la prise aiguë de drogue. Les études d’imagerie cérébrale ont montré, chez les sujets dépendants, des niveaux supra-physiologiques de dopamine dans le Nac associés à une diminution marquée de la fonction dopaminergique, avec notamment une réduction des taux de récepteurs D2 de la dopamine. Le déficit en récepteurs D2 de la dopamine pourrait jouer un rôle majeur dans la vulnérabilité à devenir dépendant. La diminution de la transmission dopaminergique est à l’origine de la baisse généralisée de la sensibilité du système de la récompense aux effets des récompenses naturelles. En revanche, les effets de la drogue et l’apprentissage conditionné entre ces effets et les stimuli neutres associés (indices contextuels) se renforcent. | Figure 6.1 Neurone dopaminergique de l’aire tegmentale ventrale projetant dans le noyau accumbens et sous le contrôle d’interneurones GABAergiques et opioïdergiques |
Circuits cérébraux de l’addiction et effets d’une exposition chronique
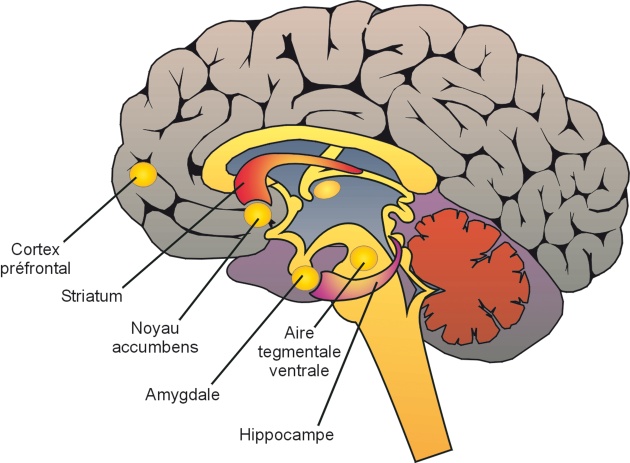
L’identification du circuit cérébral de la récompense date des années 1950 avec les travaux d’Olds et Milner (1954) : ces auteurs ont montré chez les rats que l’auto-stimulation électrique de certaines structures cérébrales (dont l’ATV) est associée à un jeûne fatal, démontrant ainsi que l’activation de ces régions cérébrales surpasse celle induite par des récompenses naturelles comme la prise de nourriture (Olds et Milner, 1954). Ainsi, l’effet des drogues se substitue à celui de récompenses naturelles pour lesquelles notre cerveau est programmé, et la drogue a un effet plus intense et prolongé. La consommation aiguë de drogue diminue le seuil de récompense (récompense augmentée) alors que la consommation chronique augmente ce seuil (récompense diminuée) et donc le besoin de consommer davantage de drogue pour atteindre ce seuil (Koob et Le Moal, 2008).
) : ces auteurs ont montré chez les rats que l’auto-stimulation électrique de certaines structures cérébrales (dont l’ATV) est associée à un jeûne fatal, démontrant ainsi que l’activation de ces régions cérébrales surpasse celle induite par des récompenses naturelles comme la prise de nourriture (Olds et Milner, 1954). Ainsi, l’effet des drogues se substitue à celui de récompenses naturelles pour lesquelles notre cerveau est programmé, et la drogue a un effet plus intense et prolongé. La consommation aiguë de drogue diminue le seuil de récompense (récompense augmentée) alors que la consommation chronique augmente ce seuil (récompense diminuée) et donc le besoin de consommer davantage de drogue pour atteindre ce seuil (Koob et Le Moal, 2008).La libération de dopamine dans le Nac joue un rôle majeur dans le développement de l’addiction. Le Nac constitue un véritable « carrefour » vers lequel convergent différentes voies de transmission en provenance de structures cérébrales variées : il reçoit des informations limbiques importantes de l’amygdale, du cortex frontal et de l’hippocampe qui sont converties en action motivée au travers de sa connectivité avec le système moteur extrapyramidal (Baler et Volkow, 2006) (figure 6.2). À côté du circuit mésolimbique (ATV et Nac), d’autres voies dopaminergiques contribuent aux effets récompensants des drogues et à l’addiction comme les voies mésostriatale (neurones dopaminergiques de la substance noire projetant dans le striatum dorsal), mésocorticale (neurones dopaminergiques de la substance noire projetant dans le cortex frontal).
) (figure 6.2). À côté du circuit mésolimbique (ATV et Nac), d’autres voies dopaminergiques contribuent aux effets récompensants des drogues et à l’addiction comme les voies mésostriatale (neurones dopaminergiques de la substance noire projetant dans le striatum dorsal), mésocorticale (neurones dopaminergiques de la substance noire projetant dans le cortex frontal).
Dans les premières étapes du développement de l’addiction, les consommations associées au plaisir voire à l’intoxication activent le circuit de la récompense (Nac, ATV et pallidum ventral) et donc celui de la sortie motrice (striatum dorsal et cortex moteur). Le circuit de la récompense est toujours sous le contrôle inhibiteur cortical du circuit impliqué dans le contrôle exécutif (cortex préfrontal dorsolatéral (CPFDL), cortex cingulaire antérieur (CCA), cortex frontal inférieur (CFI) et cortex orbitofrontal latéral (COF)). La perte de contrôle de la consommation, un des critères importants de l’addiction, se caractérise par un déséquilibre qui favorise la sur-activation des circuits de la récompense, de la motivation et de la mémoire/conditionnement (amygdale, COF médian pour l’attribution de la plus-value et le striatum dorsal pour les habitudes/automatismes) qui entraîne une exagération de la valeur attendue de la drogue (Baler et Volkow, 2006) (figure 6.3). Deux autres circuits sont aussi suractivés, impliquant un réseau neuronal jouant un rôle dans l’humeur incluant la réactivité au stress (amygdale et hypothalamus) et l’intéroception (insula et CCA) contribuant au craving. Plusieurs systèmes de neurotransmission interviennent dans ces neuro-adaptations impliquant le glutamate, le GABA, la noradrénaline, la corticolibérine (Corticotropin Releasing Factor ou CRF) et les opioïdes.
) (figure 6.3). Deux autres circuits sont aussi suractivés, impliquant un réseau neuronal jouant un rôle dans l’humeur incluant la réactivité au stress (amygdale et hypothalamus) et l’intéroception (insula et CCA) contribuant au craving. Plusieurs systèmes de neurotransmission interviennent dans ces neuro-adaptations impliquant le glutamate, le GABA, la noradrénaline, la corticolibérine (Corticotropin Releasing Factor ou CRF) et les opioïdes.

L’addiction se caractérise donc par une dérégulation de l’activité de certaines structures cérébrales, avec notamment un hypofonctionnement des régions corticales et frontales et, à l’inverse, une hyperactivation de l’amygdale. Il n’est pas clairement établi à l’heure actuelle si cette dérégulation fonctionnelle précède le développement de l’addiction (prédisposition) ou si elle est induite par la consommation chronique de drogue. La transition de l’abus vers l’addiction implique donc une augmentation de la motivation à consommer la drogue, un état émotionnel négatif et une diminution de la capacité à inhiber certains comportements (contrôle comportemental). Les lobes frontaux jouent également un rôle important dans l’impulsivité qui contribue elle aussi à la vulnérabilité à l’addiction. Il est frappant de constater que cet état d’activité de ces structures cérébrales est observé à l’état basal à l’adolescence (figure 6.4). Ceci expliquerait certains traits comportementaux (tempérament et personnalité) typiques de l’adolescence comme la recherche de sensations, la prise de risque et la plus faible capacité à planifier et à juger des conséquences de ses actes.
). Ceci expliquerait certains traits comportementaux (tempérament et personnalité) typiques de l’adolescence comme la recherche de sensations, la prise de risque et la plus faible capacité à planifier et à juger des conséquences de ses actes.

Sevrage
L’addiction se caractérise aussi par l’apparition d’un syndrome de sevrage à l’arrêt de la consommation de certaines substances et d’un état émotionnel négatif. La drogue initialement consommée pour ses effets plaisants est alors consommée, au moins en partie, pour se soulager des effets néfastes du sevrage (« renforcement négatif »). Cette phase induit une diminution de la motivation pour les récompenses naturelles alors que celle pour la drogue est largement amplifiée. Cette phase de l’addiction fait intervenir l’amygdale étendue1
jouant un rôle majeur dans le conditionnement et les émotions. Elle est composée de différents noyaux (noyau central, noyau du lit de la strie terminale et écorce du Nac). Les axes hypothalamo-hypophysaire et du stress sont recrutés et un état émotionnel négatif et une anxiété s’installent avec l’intervention de nombreux neurotransmetteurs (CRF, dynorphine, noradrénaline, neuropeptide Y, endocannabinoïdes, vasopressine et nociceptine). Le sevrage est associé à une libération accrue de CRF dans le noyau central de l’amygdale. La combinaison de l’atteinte du système de récompense et le recrutement des systèmes impliqués dans le stress constituent une base neurobiologique de l’état émotionnel négatif qui est responsable du renforcement négatif et en partie de la compulsion.
Addiction et neuroplasticité
Le stockage de la mémoire implique plusieurs formes de modification synaptique. Le principe sous-tendant ces modifications a été proposé par Donald Hebb dans son livre « The Organisation of Behavior » (Hebb, 1949). En accord avec le postulat de Hebb, une coïncidence d’activité entre deux neurones connectés synaptiquement potentialiserait l’activité synaptique entre ces deux neurones. En d’autres termes, lorsque l’axone de la cellule A est assez proche pour exciter une cellule B, provoquant de manière persistante ou répétée l’activation de cette cellule B, des processus de croissance ou de changement métabolique ont lieu dans l’une des cellules voire les deux, augmentant l’efficacité de la cellule A sur la stimulation de la cellule B. La modification de l’activité synaptique serait, selon Hebb, la base cellulaire des processus d’apprentissage. De nombreuses formes de plasticité synaptique ont été mises en évidence dans le système nerveux central (SNC). La plasticité synaptique a été définie comme un ajustement dynamique de l’efficacité ou de la force d’une synapse. Elle représente un mécanisme général par lequel les stimuli internes ou de l’environnement peuvent modifier la réponse neuronale telle que le stockage d’information acquis à travers l’expérience. La durée de ces changements synaptiques est extrêmement variable et peut aller de quelques millisecondes à plusieurs années. Il a ainsi été mis en évidence différents types de plasticité mettant en jeu des mécanismes cellulaires et moléculaires variés. On note ainsi l’existence d’une plasticité à court terme d’une durée maximale de quelques minutes, et une plasticité à long terme à partir d’une heure jusqu’à plusieurs jours. La « potentialisation à long terme » (PLT), qui se traduit par une augmentation à long terme de l’activité de la synapse et la « dépression à long terme » (DLT) qui est une diminution à long terme de l’activité de la synapse, sont des formes de plasticité qui persistent de quelques heures à plusieurs jours (Barnes et McNaughton, 1985).
). En accord avec le postulat de Hebb, une coïncidence d’activité entre deux neurones connectés synaptiquement potentialiserait l’activité synaptique entre ces deux neurones. En d’autres termes, lorsque l’axone de la cellule A est assez proche pour exciter une cellule B, provoquant de manière persistante ou répétée l’activation de cette cellule B, des processus de croissance ou de changement métabolique ont lieu dans l’une des cellules voire les deux, augmentant l’efficacité de la cellule A sur la stimulation de la cellule B. La modification de l’activité synaptique serait, selon Hebb, la base cellulaire des processus d’apprentissage. De nombreuses formes de plasticité synaptique ont été mises en évidence dans le système nerveux central (SNC). La plasticité synaptique a été définie comme un ajustement dynamique de l’efficacité ou de la force d’une synapse. Elle représente un mécanisme général par lequel les stimuli internes ou de l’environnement peuvent modifier la réponse neuronale telle que le stockage d’information acquis à travers l’expérience. La durée de ces changements synaptiques est extrêmement variable et peut aller de quelques millisecondes à plusieurs années. Il a ainsi été mis en évidence différents types de plasticité mettant en jeu des mécanismes cellulaires et moléculaires variés. On note ainsi l’existence d’une plasticité à court terme d’une durée maximale de quelques minutes, et une plasticité à long terme à partir d’une heure jusqu’à plusieurs jours. La « potentialisation à long terme » (PLT), qui se traduit par une augmentation à long terme de l’activité de la synapse et la « dépression à long terme » (DLT) qui est une diminution à long terme de l’activité de la synapse, sont des formes de plasticité qui persistent de quelques heures à plusieurs jours (Barnes et McNaughton, 1985).Les modifications persistantes du comportement, induites par des indices (« stimuli ») environnementaux ou par la consommation chronique de drogue, sont très certainement relayées par des changements durables de la transmission synaptique et de l’excitabilité neuronale, voire du nombre de connexions neuronales. Ces changements durables de la transmission synaptique ou plasticité synaptique à long terme, sont généralement définis comme une modification (augmentation/diminution ou potentialisation/dépression de l’activité) de l’efficacité de transmission au niveau d’une synapse particulière. Au niveau de l’adaptation du nombre et des caractéristiques (surface des points de contact), il s’agit d’une plasticité dite « morphologique ». Dans l’état des connaissances actuelles, ces phénomènes de plasticité apparaissent comme le meilleur substratum neurobiologique expliquant les mécanismes de l’apprentissage et de la mémorisation. De manière intéressante, les systèmes GABAergique et glutamatergique qui sont des cibles privilégiées de l’alcool, par exemple, sont aussi des acteurs essentiels des phénomènes de plasticité synaptique (Lovinger et Roberto, 2013). Relier les effets chroniques des drogues à des perturbations des mécanismes cellulaires et moléculaires à la base des processus de mémorisation conduit à la notion que les drogues sont à l’origine d’une « mémoire pathologique ». Cette mémoire pathologique explique, au moins en partie, comment les drogues laissent des traces cérébrales, ou engrammes, qui font que, même après une très longue période d’abstinence, le sujet dépendant peut rechuter lors d’une re-consommation, même faible, de drogue ou lors d’une exposition à un indice contextuel qui avait été associé avec les prises régulières de drogue.
). Relier les effets chroniques des drogues à des perturbations des mécanismes cellulaires et moléculaires à la base des processus de mémorisation conduit à la notion que les drogues sont à l’origine d’une « mémoire pathologique ». Cette mémoire pathologique explique, au moins en partie, comment les drogues laissent des traces cérébrales, ou engrammes, qui font que, même après une très longue période d’abstinence, le sujet dépendant peut rechuter lors d’une re-consommation, même faible, de drogue ou lors d’une exposition à un indice contextuel qui avait été associé avec les prises régulières de drogue.Des études précliniques récentes ont montré que la transition d’une consommation contrôlée de cocaïne vers l’addiction est liée à la perte de capacité des neurones du Nac à présenter le phénomène de « dépression à long terme » de la transmission synaptique, c’est-à-dire à présenter une diminution durable de l’efficacité de transmission synaptique (Kasanetz et coll., 2010).
).Ces adaptations d’activité synaptique (plasticité) induites par les drogues dans plusieurs régions cérébrales impliquées dans le renforcement positif ont été proposées comme étant le mécanisme cellulaire crucial qui mènerait de manière ultime à l’addiction (Kauer et Malenka, 2007). En effet, de très nombreuses études ont mis en évidence des liens solides entre comportement addictif et plasticité synaptique (Mameli et Lüscher, 2011). Par exemple, des études chez l’animal ont montré que les agents glutamatergiques qui bloquent le récepteur NMDA, un récepteur clé dans les mécanismes de plasticité synaptique, bloquent aussi différents comportements associés à l’addiction comme la préférence de place conditionnée (test mesurant les effets récompensants des drogues) ou la sensibilisation comportementale aux effets stimulants moteurs des drogues (test mesurant la tolérance inverse et l’augmentation de la motivation à consommer la drogue). De nombreux travaux ont montré une modification durable du phénomène de plasticité synaptique au niveau des synapses glutamatergiques de l’aire tegmentale ventrale (ATV) après administration aiguë ou chronique de drogues (cocaïne, amphétamine, morphine, nicotine, alcool et benzodiazépines). De manière très intéressante, cet effet est aussi observé après différents types de stress, qu’il soit aigu ou chronique (choc électrique, séparation maternelle périnatale, défaite sociale) mais il ne se produit pas lors de l’administration d’autres agents pharmacologiques comme par exemple les antidépresseurs. Au total, toutes les drogues induisent des changements persistants de la communication entre certains neurones du circuit cérébral de la récompense qui constitueraient ainsi un phénomène impliqué dans la cascade des évènements qui pourraient conduire aux comportements addictifs. Si les drogues renforcent l’efficacité de certaines synapses excitatrices au niveau des neurones dopaminergiques de l’ATV, cela modifie sûrement la libération de dopamine dans les structures cibles de l’ATV sur lesquelles elle envoie des projections, comme l’amygdale et le cortex préfrontal (CPF). Cela conduit ultimement à modifier les phénomènes d’apprentissage liés à la dopamine (Hyman et Malenka, 2001 ; Kalivas et Volkow, 2005). Même si des connaissances considérables ont été apportées durant cette dernière décennie sur les modifications de plasticité synaptique après administration de drogue, les liens de causalité entre ces phénomènes complexes et les comportements addictifs également complexes, restent à établir (Mameli et Lüscher, 2011).
). En effet, de très nombreuses études ont mis en évidence des liens solides entre comportement addictif et plasticité synaptique (Mameli et Lüscher, 2011). Par exemple, des études chez l’animal ont montré que les agents glutamatergiques qui bloquent le récepteur NMDA, un récepteur clé dans les mécanismes de plasticité synaptique, bloquent aussi différents comportements associés à l’addiction comme la préférence de place conditionnée (test mesurant les effets récompensants des drogues) ou la sensibilisation comportementale aux effets stimulants moteurs des drogues (test mesurant la tolérance inverse et l’augmentation de la motivation à consommer la drogue). De nombreux travaux ont montré une modification durable du phénomène de plasticité synaptique au niveau des synapses glutamatergiques de l’aire tegmentale ventrale (ATV) après administration aiguë ou chronique de drogues (cocaïne, amphétamine, morphine, nicotine, alcool et benzodiazépines). De manière très intéressante, cet effet est aussi observé après différents types de stress, qu’il soit aigu ou chronique (choc électrique, séparation maternelle périnatale, défaite sociale) mais il ne se produit pas lors de l’administration d’autres agents pharmacologiques comme par exemple les antidépresseurs. Au total, toutes les drogues induisent des changements persistants de la communication entre certains neurones du circuit cérébral de la récompense qui constitueraient ainsi un phénomène impliqué dans la cascade des évènements qui pourraient conduire aux comportements addictifs. Si les drogues renforcent l’efficacité de certaines synapses excitatrices au niveau des neurones dopaminergiques de l’ATV, cela modifie sûrement la libération de dopamine dans les structures cibles de l’ATV sur lesquelles elle envoie des projections, comme l’amygdale et le cortex préfrontal (CPF). Cela conduit ultimement à modifier les phénomènes d’apprentissage liés à la dopamine (Hyman et Malenka, 2001 ; Kalivas et Volkow, 2005). Même si des connaissances considérables ont été apportées durant cette dernière décennie sur les modifications de plasticité synaptique après administration de drogue, les liens de causalité entre ces phénomènes complexes et les comportements addictifs également complexes, restent à établir (Mameli et Lüscher, 2011).Adolescence et neurobiologie de l’addiction
Développement cérébral
L’adolescence est une période développementale critique qui correspond à la transition de l’enfance à l’âge adulte. Au niveau comportemental, elle se caractérise par des niveaux élevés de prise de risque, un besoin d’exploration, de nouveauté et une recherche de sensations, un niveau élevé d’interactions sociales, une activité importante, et un enclin à jouer qui sont probablement nécessaires à l’apprentissage et à l’acquisition des savoirs indispensables à la maturité et à l’indépendance vis-à-vis de la famille (Spear, 2000 ; Ernst et coll., 2009). Toutefois, la forte propension à la recherche de nouveauté et de sensations pendant cette période est aussi un facteur fortement prédictif de l’abus de substances psychoactives et du risque à développer une addiction (Baumrind, 1987 ; Andrucci et coll., 1989 ; Wills et coll., 1994 ; Faden, 2006). En effet, le cerveau adolescent présente la particularité d’être dans un état unique de transition subissant à la fois des modifications progressives et régressives procurant une base biologique pour ces comportements typiques de l’adolescence et les changements nécessaires à la maturation et à la transition vers l’âge adulte. Les études d’imagerie par résonance magnétique (IRM) chez les jeunes ont montré dès la pré-adolescence une augmentation du volume de matière grise (neurones) suivie d’une diminution à la fin de l’adolescence (Giedd et coll., 1999 ; Giedd, 2004). Au niveau cellulaire, ces changements correspondent à une myélinisation (augmentation du volume de substance blanche) et une surproduction importante de synapses pendant la phase précoce de la puberté suivie d’un élagage (suppression de connexions) dans la phase tardive de l’adolescence (Giedd et coll., 1999 ; Andersen et coll., 2000 ; Andersen et Teicher, 2004). Même si les mécanismes relayant ces changements synaptiques ne sont pas encore bien compris, il a été suggéré qu’un tel remodelage est à la base de la plasticité développementale par laquelle les circuits neuronaux s’établissent pour s’adapter aux demandes de l’environnement conduisant aux comportements adultes matures. Une telle période cruciale de remodelage rend le cerveau adolescent plus vulnérable aux agressions extérieures et aux troubles psychiatriques.
; Ernst et coll., 2009). Toutefois, la forte propension à la recherche de nouveauté et de sensations pendant cette période est aussi un facteur fortement prédictif de l’abus de substances psychoactives et du risque à développer une addiction (Baumrind, 1987 ; Andrucci et coll., 1989 ; Wills et coll., 1994 ; Faden, 2006). En effet, le cerveau adolescent présente la particularité d’être dans un état unique de transition subissant à la fois des modifications progressives et régressives procurant une base biologique pour ces comportements typiques de l’adolescence et les changements nécessaires à la maturation et à la transition vers l’âge adulte. Les études d’imagerie par résonance magnétique (IRM) chez les jeunes ont montré dès la pré-adolescence une augmentation du volume de matière grise (neurones) suivie d’une diminution à la fin de l’adolescence (Giedd et coll., 1999 ; Giedd, 2004). Au niveau cellulaire, ces changements correspondent à une myélinisation (augmentation du volume de substance blanche) et une surproduction importante de synapses pendant la phase précoce de la puberté suivie d’un élagage (suppression de connexions) dans la phase tardive de l’adolescence (Giedd et coll., 1999 ; Andersen et coll., 2000 ; Andersen et Teicher, 2004). Même si les mécanismes relayant ces changements synaptiques ne sont pas encore bien compris, il a été suggéré qu’un tel remodelage est à la base de la plasticité développementale par laquelle les circuits neuronaux s’établissent pour s’adapter aux demandes de l’environnement conduisant aux comportements adultes matures. Une telle période cruciale de remodelage rend le cerveau adolescent plus vulnérable aux agressions extérieures et aux troubles psychiatriques.Le cortex préfrontal (CPF) et le système limbique qui comprend l’hippocampe, l’amygdale, le Nac et l’hypothalamus, subissent une réorganisation importante pendant l’adolescence. En effet, le volume de matière grise diminue chez l’Homme (Sowell et coll., 1999 et 2001) et chez l’animal (van Eden et coll., 1990) en fin d’adolescence. Parallèlement, une perte substantielle de synapses, en particulier les synapses excitatrices glutamatergiques du CPF, est observée (Huttenlocher, 1984 ; Zecevic et coll., 1989). À l’inverse de ce phénomène d’élagage, les connexions dopaminergiques et sérotoninergiques augmentent dans le CPF (Kalsbeek et coll., 1988 ; Rosenberg et Lewis, 1994). De la même manière, l’innervation cholinergique du CPF augmente (Kostovic, 1990). Au niveau de l’hippocampe, de l’amygdale, du Nac et de l’hypothalamus, il se produit un élagage important du nombre de connexions synaptiques contribuant à un remodelage morphologique. L’ensemble de ces modifications caractérise la maturation des aires corticales et limbiques à l’adolescence.
et 2001) et chez l’animal (van Eden et coll., 1990) en fin d’adolescence. Parallèlement, une perte substantielle de synapses, en particulier les synapses excitatrices glutamatergiques du CPF, est observée (Huttenlocher, 1984 ; Zecevic et coll., 1989). À l’inverse de ce phénomène d’élagage, les connexions dopaminergiques et sérotoninergiques augmentent dans le CPF (Kalsbeek et coll., 1988 ; Rosenberg et Lewis, 1994). De la même manière, l’innervation cholinergique du CPF augmente (Kostovic, 1990). Au niveau de l’hippocampe, de l’amygdale, du Nac et de l’hypothalamus, il se produit un élagage important du nombre de connexions synaptiques contribuant à un remodelage morphologique. L’ensemble de ces modifications caractérise la maturation des aires corticales et limbiques à l’adolescence.Les études comportementales ont démontré une évolution des performances dans des tâches impliquant le contrôle inhibiteur, la prise de décision et la vitesse de traitement de l’information pendant l’adolescence. L’attention sélective, la mémoire de travail et la capacité de résolution de problème s’améliorent progressivement au cours de cette période, parallèlement à la progression de la myélinisation et l’élagage synaptique fronto-cortical (Blakemore et Choudhury, 2006). De manière similaire, le contrôle inhibiteur exécutif s’améliore de l’adolescence à l’âge adulte. Les études analysant l’inhibition comportementale avec une tâche de Go-No Go2
et utilisant l’IRM fonctionnelle révèlent une plus grande activation des cortex orbitofrontal et dorsolatéral chez les enfants que chez les adolescents ; cette activation chez les adolescents est supérieure à celle observée chez les adultes, ces derniers présentant la plus faible activation dorsolatérale, mais une activation orbitofrontale identique et une performance accrue du contrôle inhibiteur (Casey et coll., 1997 ; Tamm et coll., 2002). Ces études ont permis d’établir le concept selon lequel le cerveau immature, avec un excès de synapses, présente une plus grande activation corticale (mais moins efficiente) et une plus faible performance que celui des adultes ; ces derniers ont quant-à-eux un cortex frontal plus efficient, avec pour conséquences une activation plus focalisée et moins importante globalement, un temps de réaction plus rapide et de meilleures performances (Blakemore et Choudhury, 2006). Dans leur globalité, ces études suggèrent que le remodelage du cortex pendant la période de transition entre l’adolescence et l’âge adulte exerce un rôle fonctionnel crucial sur le devenir à l’âge adulte.
). De manière similaire, le contrôle inhibiteur exécutif s’améliore de l’adolescence à l’âge adulte. Les études analysant l’inhibition comportementale avec une tâche de Go-No Go2
et utilisant l’IRM fonctionnelle révèlent une plus grande activation des cortex orbitofrontal et dorsolatéral chez les enfants que chez les adolescents ; cette activation chez les adolescents est supérieure à celle observée chez les adultes, ces derniers présentant la plus faible activation dorsolatérale, mais une activation orbitofrontale identique et une performance accrue du contrôle inhibiteur (Casey et coll., 1997 ; Tamm et coll., 2002). Ces études ont permis d’établir le concept selon lequel le cerveau immature, avec un excès de synapses, présente une plus grande activation corticale (mais moins efficiente) et une plus faible performance que celui des adultes ; ces derniers ont quant-à-eux un cortex frontal plus efficient, avec pour conséquences une activation plus focalisée et moins importante globalement, un temps de réaction plus rapide et de meilleures performances (Blakemore et Choudhury, 2006). Dans leur globalité, ces études suggèrent que le remodelage du cortex pendant la période de transition entre l’adolescence et l’âge adulte exerce un rôle fonctionnel crucial sur le devenir à l’âge adulte.Sensibilité à la récompense
La propension des adolescents à la recherche de sensations et à la prise de risque ainsi que leur faible capacité à la prise de décision font qu’ils sont plus sensibles aux récompenses. Deux théories ont été avancées pour expliquer cette plus forte sensibilité à la récompense. La première postule que l’hypoactivation du striatum pousse les adolescents à s’engager dans un comportement de recherche de récompense comme une réponse compensatoire, ou « d’auto-médication ». La seconde théorie suggère, au contraire, que c’est l’hyperactivation du striatum qui induit un comportement de recherche de récompense. Les études récentes d’imagerie cérébrale soutiennent plutôt cette dernière hypothèse (Galvan, 2010). En effet, une plus grande activation du striatum ventral (Nac) a été démontrée chez les adolescents comparativement aux enfants et aux adultes lors de l’anticipation d’une récompense (Galvan et coll., 2006 ; Geier et coll., 2010) ou de la réception de cette récompense (Van Leijenhorst et coll., 2010). Durant le traitement de la récompense, le striatum ventral présente une diminution d’activité quand les adolescents doivent évaluer la valeur d’une récompense, mais une augmentation d’activité quand ils anticipent la récompense (Geier et coll., 2010). Ces observations suggèrent chez les adolescents comparativement aux adultes, des capacités limitées à évaluer la valeur de la récompense (correspondant à la diminution d’activité du striatum ventral), associées à une réactivité exagérée lors de l’anticipation de la récompense. L’hyperactivation du striatum peut être associée à une augmentation de la libération de dopamine dans le striatum ventral (Koepp et coll., 1998 ; Aarts et coll., 2010). Une plus grande libération de dopamine pourrait donc expliquer pourquoi les adolescents sont plus en recherche de récompense, créant ainsi un cycle de renforcement de ce comportement.
). En effet, une plus grande activation du striatum ventral (Nac) a été démontrée chez les adolescents comparativement aux enfants et aux adultes lors de l’anticipation d’une récompense (Galvan et coll., 2006 ; Geier et coll., 2010) ou de la réception de cette récompense (Van Leijenhorst et coll., 2010). Durant le traitement de la récompense, le striatum ventral présente une diminution d’activité quand les adolescents doivent évaluer la valeur d’une récompense, mais une augmentation d’activité quand ils anticipent la récompense (Geier et coll., 2010). Ces observations suggèrent chez les adolescents comparativement aux adultes, des capacités limitées à évaluer la valeur de la récompense (correspondant à la diminution d’activité du striatum ventral), associées à une réactivité exagérée lors de l’anticipation de la récompense. L’hyperactivation du striatum peut être associée à une augmentation de la libération de dopamine dans le striatum ventral (Koepp et coll., 1998 ; Aarts et coll., 2010). Une plus grande libération de dopamine pourrait donc expliquer pourquoi les adolescents sont plus en recherche de récompense, créant ainsi un cycle de renforcement de ce comportement.Les processus de maturation durant la puberté sont associés à l’augmentation de la recherche de récompense (Galvan et coll., 2007) et joueraient un rôle dans la sensibilité à cette récompense. Forbes et coll. (2010) ont mis en évidence une moindre activation striatale et une plus forte activation du cortex préfrontal médian en réponse à une récompense financière (gain, perte ou pas de changement) chez les adolescents présentant une maturation pubertaire plus avancée que chez ceux du même âge avec une maturation moins avancée. En outre, le rôle potentiel du cortex préfrontal médian dans la cognition sociale et les représentations personnelles suggère que les adolescents en pleine phase de maturation attachent une importance particulière au contexte social et à l’influence des pairs quand ils répondent à une récompense (Forbes et coll., 2010). L’état affectif interagirait aussi avec la réponse neuronale à la récompense puisqu’une activité faible et élevée, respectivement du striatum et du cortex préfrontal, a été associée à des symptômes dépressifs. En effet, les adolescents présentent un risque accru de comportement à risque lorsque la situation nécessite un traitement affectif. Les niveaux hormonaux joueraient aussi un rôle dans la sensibilité à la récompense. Des niveaux élevés de testostérone ont été associés avec une activité striatale réduite en réponse à une récompense à la fois chez les filles et chez les garçons. En raison de ce fonctionnement cérébral particulier, les adolescents pourraient donc présenter une plus grande propension et un intérêt accru pour les récompenses qui les poussent à prendre des risques et à rechercher des sensations (Martin et coll., 2004 ; Forbes et coll., 2010).
) et joueraient un rôle dans la sensibilité à cette récompense. Forbes et coll. (2010) ont mis en évidence une moindre activation striatale et une plus forte activation du cortex préfrontal médian en réponse à une récompense financière (gain, perte ou pas de changement) chez les adolescents présentant une maturation pubertaire plus avancée que chez ceux du même âge avec une maturation moins avancée. En outre, le rôle potentiel du cortex préfrontal médian dans la cognition sociale et les représentations personnelles suggère que les adolescents en pleine phase de maturation attachent une importance particulière au contexte social et à l’influence des pairs quand ils répondent à une récompense (Forbes et coll., 2010). L’état affectif interagirait aussi avec la réponse neuronale à la récompense puisqu’une activité faible et élevée, respectivement du striatum et du cortex préfrontal, a été associée à des symptômes dépressifs. En effet, les adolescents présentent un risque accru de comportement à risque lorsque la situation nécessite un traitement affectif. Les niveaux hormonaux joueraient aussi un rôle dans la sensibilité à la récompense. Des niveaux élevés de testostérone ont été associés avec une activité striatale réduite en réponse à une récompense à la fois chez les filles et chez les garçons. En raison de ce fonctionnement cérébral particulier, les adolescents pourraient donc présenter une plus grande propension et un intérêt accru pour les récompenses qui les poussent à prendre des risques et à rechercher des sensations (Martin et coll., 2004 ; Forbes et coll., 2010).Génétique et addiction
Tous les individus ne sont pas égaux en termes de vulnérabilité à développer une addiction, laquelle dépend de l’interaction entre des facteurs génétiques et environnementaux. Les facteurs génétiques jouent un rôle capital puisqu’ils contribueraient à hauteur d’environ 50 % de la variance du risque à développer la maladie.
En effet, approximativement 40 à 60 % du risque (ou plus précisément de la variance du risque) de développer une addiction seraient attribuables à des facteurs génétiques (Prescott et Kendler, 1999 ; Goldman et coll., 2005). Par exemple, des études d’agrégation familiale apportent la preuve d’une forte héritabilité de l’addiction à l’alcool (Bierut et coll., 1998 ; Merikangas et coll., 1998). Les études d’adoption ont aussi révélé que l’addiction à l’alcool chez les adoptés était liée à celle des parents biologiques et non à celle des parents d’adoption (Cloninger et coll., 1981). Cependant, la majorité des connaissances sur l’héritabilité de l’addiction à l’alcool provient des études de concordances des jumeaux monozygotes comparativement aux dizygotes (Viken et coll., 1999 ; Hopfer et coll., 2003). Le degré de similitude entre jumeaux, pour des traits quantitatifs comme l’addiction, s’exprime par un taux de concordance. Pour qu’un trouble donné puisse être considéré comme lié uniquement à des facteurs génétiques, il faudrait que ce taux soit égal à 100 % chez les jumeaux monozygotes. Les nombreuses études familiales, d’adoption et de jumeaux ont permis de déterminer l’importance des facteurs héritables concernant les différences individuelles dans l’addiction. Les résultats des études de jumeaux ont ainsi établi que le pourcentage de la variance dans la vulnérabilité est de 71 % pour la dépendance à la nicotine, 48-66 % pour la dépendance à l’alcool, 51-59% pour la dépendance au cannabis, 42-79 % pour la dépendance à la cocaïne, 23-54 % pour la dépendance aux opiacés et 49 % pour le jeu pathologique (voir pour revue Agrawal et coll., 2012).
; Goldman et coll., 2005). Par exemple, des études d’agrégation familiale apportent la preuve d’une forte héritabilité de l’addiction à l’alcool (Bierut et coll., 1998 ; Merikangas et coll., 1998). Les études d’adoption ont aussi révélé que l’addiction à l’alcool chez les adoptés était liée à celle des parents biologiques et non à celle des parents d’adoption (Cloninger et coll., 1981). Cependant, la majorité des connaissances sur l’héritabilité de l’addiction à l’alcool provient des études de concordances des jumeaux monozygotes comparativement aux dizygotes (Viken et coll., 1999 ; Hopfer et coll., 2003). Le degré de similitude entre jumeaux, pour des traits quantitatifs comme l’addiction, s’exprime par un taux de concordance. Pour qu’un trouble donné puisse être considéré comme lié uniquement à des facteurs génétiques, il faudrait que ce taux soit égal à 100 % chez les jumeaux monozygotes. Les nombreuses études familiales, d’adoption et de jumeaux ont permis de déterminer l’importance des facteurs héritables concernant les différences individuelles dans l’addiction. Les résultats des études de jumeaux ont ainsi établi que le pourcentage de la variance dans la vulnérabilité est de 71 % pour la dépendance à la nicotine, 48-66 % pour la dépendance à l’alcool, 51-59% pour la dépendance au cannabis, 42-79 % pour la dépendance à la cocaïne, 23-54 % pour la dépendance aux opiacés et 49 % pour le jeu pathologique (voir pour revue Agrawal et coll., 2012).Le risque de développer une addiction à l’alcool est multiplié par 4 à 5 chez les enfants de parents alcoolo-dépendants (Goodwin et coll., 1974 ; Schuckit, 1995). Les enfants de parents alcoolo-dépendants sont plus sensibles aux effets récompensants de l’alcool lors de la phase ascendante du pic d’alcoolémie et moins sensibles aux effets sédatifs/hypnotiques et à l’incoordination motrice lors de la phase descendante du pic d’alcoolémie. Des études sur des familles sévèrement touchées par l’addiction à l’alcool ont montré qu’environ 50 % des frères et 22-25 % des sœurs de sujets alcoolo-dépendants sont également alcoolo-dépendants (Bierut et coll., 1998). De la même manière, les frères et sœurs de sujets dépendants au cannabis, à la cocaïne ou à la nicotine ont un risque accru (environ 1,7 fois) de développer un dépendance au cannabis, à la cocaïne ou à la nicotine comparativement aux sujets d’une fratrie sans sujet dépendant (Bierut et coll., 1998). Par ailleurs, il est maintenant clairement établi que la susceptibilité génétique à l’alcoolo-dépendance est gouvernée par plusieurs gènes (susceptibilité polygénique) qui, pris séparément, n’ont qu’un effet modeste sur le phénotype global exprimé (Merikangas et Risch, 2003).
; Schuckit, 1995). Les enfants de parents alcoolo-dépendants sont plus sensibles aux effets récompensants de l’alcool lors de la phase ascendante du pic d’alcoolémie et moins sensibles aux effets sédatifs/hypnotiques et à l’incoordination motrice lors de la phase descendante du pic d’alcoolémie. Des études sur des familles sévèrement touchées par l’addiction à l’alcool ont montré qu’environ 50 % des frères et 22-25 % des sœurs de sujets alcoolo-dépendants sont également alcoolo-dépendants (Bierut et coll., 1998). De la même manière, les frères et sœurs de sujets dépendants au cannabis, à la cocaïne ou à la nicotine ont un risque accru (environ 1,7 fois) de développer un dépendance au cannabis, à la cocaïne ou à la nicotine comparativement aux sujets d’une fratrie sans sujet dépendant (Bierut et coll., 1998). Par ailleurs, il est maintenant clairement établi que la susceptibilité génétique à l’alcoolo-dépendance est gouvernée par plusieurs gènes (susceptibilité polygénique) qui, pris séparément, n’ont qu’un effet modeste sur le phénotype global exprimé (Merikangas et Risch, 2003).Concernant la nicotine, une récente méta-analyse des études de jumeaux a montré que les facteurs génétiques et environnementaux influencent la propension à fumer (Li et Burmeister, 2009). Ainsi, chez les femmes, l’initiation de l’usage de nicotine est largement influencée par les facteurs génétiques, alors que chez les hommes l’impact génétique porte davantage sur le maintien de l’usage que sur l’initiation. L’héritabilité de l’initiation et de la dépendance à la nicotine est estimée respectivement à 50 % et 59 %. Une méta-analyse des études de jumeaux sur le cannabis a montré que la vulnérabilité à initier la consommation et à un usage répété dépend elle aussi de facteurs génétiques et environnementaux (Verweij et coll., 2010). Ainsi les facteurs génétiques ont compté pour 48 % et 40 % de la variance totale à initier la consommation respectivement chez les hommes et les femmes, et 51 % et 59 % de la variance totale à l’usage problématique respectivement chez les hommes et les femmes.
). Ainsi, chez les femmes, l’initiation de l’usage de nicotine est largement influencée par les facteurs génétiques, alors que chez les hommes l’impact génétique porte davantage sur le maintien de l’usage que sur l’initiation. L’héritabilité de l’initiation et de la dépendance à la nicotine est estimée respectivement à 50 % et 59 %. Une méta-analyse des études de jumeaux sur le cannabis a montré que la vulnérabilité à initier la consommation et à un usage répété dépend elle aussi de facteurs génétiques et environnementaux (Verweij et coll., 2010). Ainsi les facteurs génétiques ont compté pour 48 % et 40 % de la variance totale à initier la consommation respectivement chez les hommes et les femmes, et 51 % et 59 % de la variance totale à l’usage problématique respectivement chez les hommes et les femmes.Des études sur l’étiologie de la comorbidité (co-occurrence) de l’usage de différentes drogues (polyconsommation) chez les adolescents ont suggéré l’existence d’influences génétiques et environnementales communes entre les différentes drogues (Rhee et coll., 2003 ; Kendler et coll., 2008).
; Kendler et coll., 2008).De nombreux polymorphismes de gènes ont été associés avec le risque de développer une addiction ou bien encore de présenter un phénotype sévère de la maladie. Ces gènes candidats concernent principalement des enzymes et des récepteurs impliqués dans les mécanismes de transmission neuronale ou des voies de signalisation cellulaire. Cette vulnérabilité génétique est aussi très largement démontrée grâce aux modèles animaux. Différentes souches de rats ou de souris présentent une appétence quasi nulle ou à l’inverse extrême vis-à-vis de l’alcool par exemple. Il a également été possible de sélectionner génétiquement des animaux pour leur appétence pour l’alcool (Bell et coll., 2006 ; Naassila, 2013).
; Naassila, 2013).La réponse subjective aux effets de l’alcool peut dépendre de la mutation d’une seule base au niveau de l’ADN, comme celle du gène codant le récepteur mu des opioïdes endogènes (OPRM1). L’alcool induit dans le cerveau une libération d’opioïdes qui est impliquée dans ses effets plaisants. L’étude de Ramchandani et coll. (2011) a démontré une sensibilité accrue aux effets de l’alcool et une plus forte libération de dopamine dans le circuit de la récompense chez les sujets masculins porteurs de l’allèle polymorphe 118G d’OPRM1.
) a démontré une sensibilité accrue aux effets de l’alcool et une plus forte libération de dopamine dans le circuit de la récompense chez les sujets masculins porteurs de l’allèle polymorphe 118G d’OPRM1.Les études d’association génétique ont mis en évidence des gènes candidats impliqués dans différents systèmes de neurotransmission, notamment sérotoninergique, dopaminergique, GABAergique et cholinergique. De nombreuses études ont permis d’associer certains polymorphismes de ces gènes candidats à l’abus de drogues et à l’addiction (Wang et coll. 2012). Alors que certains polymorphismes sont communs aux différents types de comportements addictifs, certains ont été identifiés comme étant plus spécifiques. Dans l’addiction à l’alcool, on retrouve notamment les gènes : ADH, ALDH2, DRD1, DRD2/ANKK1, DRD3, DRD4, DAT, MAOA, COMT, GABRA2, SLC6A4 (Gorwood et coll., 2012) ; pour l’addiction aux stimulants : DBH, CNR1, DRD2/ANKK1, NCAM1, TTC12, CALCYON ; pour le tabac, on retrouve les gènes codant les sous-unités du récepteur nicotinique de l’acétylcholine : CHRNA5, CHRNA3, CHRNB4, CHRNA, CHRNB3 ; pour les opiacés : OPRM1 ; pour le cannabis : CNR1, FAAH ; pour le jeu pathologique : DRD1.
). Alors que certains polymorphismes sont communs aux différents types de comportements addictifs, certains ont été identifiés comme étant plus spécifiques. Dans l’addiction à l’alcool, on retrouve notamment les gènes : ADH, ALDH2, DRD1, DRD2/ANKK1, DRD3, DRD4, DAT, MAOA, COMT, GABRA2, SLC6A4 (Gorwood et coll., 2012) ; pour l’addiction aux stimulants : DBH, CNR1, DRD2/ANKK1, NCAM1, TTC12, CALCYON ; pour le tabac, on retrouve les gènes codant les sous-unités du récepteur nicotinique de l’acétylcholine : CHRNA5, CHRNA3, CHRNB4, CHRNA, CHRNB3 ; pour les opiacés : OPRM1 ; pour le cannabis : CNR1, FAAH ; pour le jeu pathologique : DRD1.La variation interindividuelle dans la motivation à consommer résulterait de réponses neurobiologiques différentes à l’alcool (renforcement ou sédation) qui sont dépendantes de facteurs génétiques. Pour l’alcool, il a aussi été suggéré que les attentes et les motivations à consommer le produit pourraient en partie être associées à des facteurs génétiques, pouvant conduire à une consommation à risque (Prescott et coll., 2004). Les facteurs génétiques joueraient un rôle important pendant les premières phases de l’adolescence, puis ensuite les facteurs environnementaux, notamment la pression du groupe de pairs, gagneraient en importance.
). Les facteurs génétiques joueraient un rôle important pendant les premières phases de l’adolescence, puis ensuite les facteurs environnementaux, notamment la pression du groupe de pairs, gagneraient en importance.Très peu de ces gènes ont été associés à la consommation à risque et au binge drinking, notamment chez les adolescents et les jeunes adultes. Pourtant, Viken et coll. (1999) ont montré par exemple que 56 % de la variance dans la fréquence des consommations jusqu’à intoxication chez des jeunes âgés de 17 ans, était expliquée par des facteurs génétiques. Treutlein et coll. (2006) ont trouvé une association significative entre deux haplotypes tag-SNP (htSNPs)3
du récepteur de type 1 du CRH (Corticotropin Releasing Hormone) et la pratique du binge drinking, la prévalence vie entière d’usage d’alcool et d’intoxication alcoolique. Une autre association a été observée entre un polymorphisme du promoteur du gène codant le transporteur de la sérotonine 5-HTTLPR (SLC6A4) et la fréquence du binge drinking, la fréquence de la consommation jusqu’à intoxication et un nombre plus élevé de verres consommés par occasion chez les étudiants (Herman et coll., 2003). Un polymorphisme (nombre variable de répétitions en tandem de 48 bases) dans l’exon III du gène DRD4 du récepteur dopaminergique D4 a été montré comme étant associé au binge drinking chez les jeunes de 18 à 23 ans (Vaughn et coll., 2009).
) ont montré par exemple que 56 % de la variance dans la fréquence des consommations jusqu’à intoxication chez des jeunes âgés de 17 ans, était expliquée par des facteurs génétiques. Treutlein et coll. (2006) ont trouvé une association significative entre deux haplotypes tag-SNP (htSNPs)3
du récepteur de type 1 du CRH (Corticotropin Releasing Hormone) et la pratique du binge drinking, la prévalence vie entière d’usage d’alcool et d’intoxication alcoolique. Une autre association a été observée entre un polymorphisme du promoteur du gène codant le transporteur de la sérotonine 5-HTTLPR (SLC6A4) et la fréquence du binge drinking, la fréquence de la consommation jusqu’à intoxication et un nombre plus élevé de verres consommés par occasion chez les étudiants (Herman et coll., 2003). Un polymorphisme (nombre variable de répétitions en tandem de 48 bases) dans l’exon III du gène DRD4 du récepteur dopaminergique D4 a été montré comme étant associé au binge drinking chez les jeunes de 18 à 23 ans (Vaughn et coll., 2009).Le gène du transporteur de la sérotonine, SLC6A4 (5-HTTLPR) présente dans sa région promotrice un polymorphisme fonctionnel modifiant l’activité transcriptionnelle. Comparé au variant « long » (L), l’allèle « court » (S) induit une plus faible activité transcriptionnelle du gène in vitro. L’allèle S est associé à une plus faible expression du transporteur et à une moindre recapture de la sérotonine. Une méta-analyse de 13 populations indépendantes a montré une association avec l’alcoolo-dépendance. Des études ont montré que : 1) les sujets alcoolo-dépendants porteurs de l’allèle L présentent un craving plus intense ; 2) la consommation excessive d’alcool et la plus faible réponse aux effets de l’alcool sont observées chez les porteurs de l’allèle L ; 3) une proportion plus importante de binge drinkers chez les étudiants homozygotes pour l’allèle S (Herman et coll., 2003 ; voir aussi Olsson et coll., 2005).
; voir aussi Olsson et coll., 2005).Le système dopaminergique, avec notamment les récepteurs D1, D2, D3 et D4 de la dopamine et la catéchol-O-méthyltransférase (COMT), joue un rôle majeur dans le métabolisme de la dopamine et de la noradrénaline ; il a été beaucoup étudié dans le cadre de la consommation excessive d’alcool et de l’alcoolo-dépendance. Le système dopaminergique mésolimbique, avec les projections dopaminergiques de l’ATV vers le Nac et les autres structures du cerveau antérieur, est impliqué dans le développement et la maintenance des maladies addictives dont l’addiction à l’alcool. Ce système est modulé par des afférences excitatrices et inhibitrices, incluant les neurones glutamatergiques et GABAergiques ainsi que par les opioïdes endogènes. L’activation des récepteurs GABAA joue un rôle important dans les réponses comportementales à l’alcool incluant les réponses motrices, anxiolytiques et sédatives. Parmi les gènes candidats, la sous-unité alpha 2 du récepteur GABAA (GABRA2) a été associée à l’alcoolo-dépendance (Edenberg et coll., 2004). Dans une étude récente d’imagerie, le polymorphisme SNP (rs279871) du gène GABRA2 a été associé à une plus forte activation du cortex fronto-médian et de l’ATV en réponse à l’odeur de la boisson alcoolisée préférée (Kareken et coll., 2010).
). Dans une étude récente d’imagerie, le polymorphisme SNP (rs279871) du gène GABRA2 a été associé à une plus forte activation du cortex fronto-médian et de l’ATV en réponse à l’odeur de la boisson alcoolisée préférée (Kareken et coll., 2010).Concernant le récepteur D1, il a été mis en évidence que l’allèle T présentant le polymorphisme SNP rs686, était associé à la sévérité de l’alcoolo-dépendance (Batel et coll., 2008).
).Le gène codant le récepteur D2 de la dopamine (DRD2) a été l’un des premiers gènes candidats identifiés dans les études d’association avec l’addiction à l’alcool. L’allèle DRD2 Taq1A A1 (rs1800497) est associé à une diminution du taux et de la fonctionnalité du récepteur D2, ce qui pourrait expliquer au moins en partie la consommation d’alcool à visée d’automédication pour compenser ce déficit, l’alcool augmentant la libération de dopamine. De nombreuses études ont associé ce polymorphisme de DRD2 à l’usage d’alcool, la dépendance et sa sévérité (Blum et coll., 1993). De manière intéressante, une étude a montré que les adolescents qui consomment de l’alcool pour faire face à leurs problèmes ou pour soulager leurs sentiments négatifs, présentent des niveaux de binge drinking et de problèmes liés à l’alcool plus élevés que ceux qui boivent pour d’autres raisons, et ce d’autant plus s’ils sont porteurs de l’allèle DRD2 A1 (van der Zwaluw et coll., 2011). Le variant DRD3 Ser9Gly (rs6280) a quant à lui été associé à l’alcoolo-dépendance (Limosin et coll., 2005).
). De manière intéressante, une étude a montré que les adolescents qui consomment de l’alcool pour faire face à leurs problèmes ou pour soulager leurs sentiments négatifs, présentent des niveaux de binge drinking et de problèmes liés à l’alcool plus élevés que ceux qui boivent pour d’autres raisons, et ce d’autant plus s’ils sont porteurs de l’allèle DRD2 A1 (van der Zwaluw et coll., 2011). Le variant DRD3 Ser9Gly (rs6280) a quant à lui été associé à l’alcoolo-dépendance (Limosin et coll., 2005).Des gènes candidats jouant un rôle dans d’autres systèmes de neurotransmission ont aussi été décrits. Le système opioïdergique endogène joue également un rôle crucial dans la physiopathologie de l’addiction à l’alcool. La consommation d’alcool augmente l’activité opioïdergique qui lève l’inhibition GABAergique sur les neurones dopaminergiques du circuit cérébral de la récompense, entraînant la libération de dopamine impliquée dans les effets renforçants de l’alcool. Étant donné le rôle essentiel du système opioïdergique dans les effets pharmacologiques de l’alcool, les études d’association génétique ont permis d’établir que le gène codant le récepteur mu des opioïdes OPRM1 était un gène candidat dans l’addiction à l’alcool. Une étude récente s’intéressant au polymorphisme fonctionnel A118G OPRM1 (rs1799971) a montré que les adolescents porteurs de cet allèle G consommaient de l’alcool pour ses effets positifs, plaisants qui sont davantage ressentis, comparativement aux homozygotes pour l’allèle A : cette augmentation de la sensation des effets plaisants est impliquée dans l’association entre le génotype OPRM1 et les problèmes liés à la consommation d’alcool. Des polymorphismes du gène codant le récepteur cholinergique muscarinique de type 2 ont aussi été associés à l’alcoolo-dépendance ou à sa sévérité. Les résultats récents du projet européen IMAGEN mené chez les adolescents ont montré l’influence d’un polymorphisme dans le gène codant la sous-unité alpha3 du récepteur nicotinique sur la vulnérabilité à l’addiction à la nicotine : la présence de ce polymorphisme perturberait l’activité du réseau mésocorticolimbique et modifierait ainsi la capacité des individus à adapter leur comportement face à une récompense et la sensibilité à la récompense serait réduite (Nees et coll., 2013). Ce même consortium de recherche a aussi révélé l’effet d’un polymorphisme du gène codant RASGRF2 (une molécule de signalisation intracellulaire) sur la sensibilité à la récompense chez les adolescents : ce polymorphisme est associé à une plus forte fréquence de consommation abusive d’alcool qui serait en lien avec une dérégulation de l’activité des neurones dopaminergiques du circuit de la récompense (Stacey et coll., 2012).
). Ce même consortium de recherche a aussi révélé l’effet d’un polymorphisme du gène codant RASGRF2 (une molécule de signalisation intracellulaire) sur la sensibilité à la récompense chez les adolescents : ce polymorphisme est associé à une plus forte fréquence de consommation abusive d’alcool qui serait en lien avec une dérégulation de l’activité des neurones dopaminergiques du circuit de la récompense (Stacey et coll., 2012).Les gènes identifiés comme étant impliqués dans les addictions aux substances psychoactives jouent un rôle à différents niveaux. Ils interviennent par exemple dans le métabolisme de la substance (métabolisme de l’alcool : alcool déshydrogénase, acétaldéhyde déshydrogénase), la sensibilité aux effets plaisants/récompensants, ou encore la sévérité de l’addiction. D’autres gènes ont été impliqués dans des comportements/traits phénotypiques (encore appelés « endophénotypes » qui sont des phénotypes intermédiaires plus restreints) comme la désinhibition, l’attention, certaines activités cérébrales (encéphalogramme), la réponse au stress, la recherche de sensations ou l’impulsivité.
Relativement peu d’études ont analysé la contribution respective des facteurs génétiques et environnementaux à l’adolescence susceptibles d’influer sur l’initiation de la consommation, la fréquence des consommations et l’apparition des problèmes liés à la consommation. Pagan et coll. (2006) rapportent dans une étude de jumeaux que les facteurs environnementaux jouent un rôle prépondérant dans l’initiation de la consommation d’alcool à l’adolescence et un rôle moindre relativement à la fréquence des consommations et aux dommages associés chez les jeunes adultes (25 ans) où les facteurs génétiques semblent intervenir de manière plus importante. Cette étude confirme les résultats obtenus précédemment par d’autres, toujours chez les jumeaux, montrant que l’initiation de la consommation d’alcool est largement influencée par des facteurs de risque environnementaux comme des facteurs socio-régionaux, l’interaction avec la fratrie, le profil de consommation des parents (Kaprio et coll., 1987 ; Heath et Martin, 1988 ; Heath et coll., 1991 ; Koopmans et Boomsma, 1996 ; Stallings et coll., 1999 ; Rose et coll., 2001). Par exemple, Rose et coll. (2001) ont estimé que les facteurs environnementaux participent à hauteur de 76 % dans la variance du risqué à initier une consommation d’alcool chez les garçons et les filles. Dans une revue de 18 études, Hopfer et coll. (2003) ont trouvé que l’initiation de la consommation d’alcool est sous l’influence de facteurs environnementaux à raison de 55-80% de la variance. Une fois la consommation initiée, les facteurs génétiques expliqueraient pour une large part la variation dans la fréquence de l’usage d’alcool (34-72 %), spécialement avec l’avancée en âge des adolescents (Heath et coll., 1991 ; Koopsmans et Boomsma, 1996 ; Maes et coll., 1999 ; Viken et coll., 1999 ; Rose et coll., 2001 ; Hopfer et coll., 2003). De plus, la rapidité dans la transition du premier usage à une consommation régulière est également largement influencée par des facteurs génétiques (Stallings et coll., 1999).
) rapportent dans une étude de jumeaux que les facteurs environnementaux jouent un rôle prépondérant dans l’initiation de la consommation d’alcool à l’adolescence et un rôle moindre relativement à la fréquence des consommations et aux dommages associés chez les jeunes adultes (25 ans) où les facteurs génétiques semblent intervenir de manière plus importante. Cette étude confirme les résultats obtenus précédemment par d’autres, toujours chez les jumeaux, montrant que l’initiation de la consommation d’alcool est largement influencée par des facteurs de risque environnementaux comme des facteurs socio-régionaux, l’interaction avec la fratrie, le profil de consommation des parents (Kaprio et coll., 1987 ; Heath et Martin, 1988 ; Heath et coll., 1991 ; Koopmans et Boomsma, 1996 ; Stallings et coll., 1999 ; Rose et coll., 2001). Par exemple, Rose et coll. (2001) ont estimé que les facteurs environnementaux participent à hauteur de 76 % dans la variance du risqué à initier une consommation d’alcool chez les garçons et les filles. Dans une revue de 18 études, Hopfer et coll. (2003) ont trouvé que l’initiation de la consommation d’alcool est sous l’influence de facteurs environnementaux à raison de 55-80% de la variance. Une fois la consommation initiée, les facteurs génétiques expliqueraient pour une large part la variation dans la fréquence de l’usage d’alcool (34-72 %), spécialement avec l’avancée en âge des adolescents (Heath et coll., 1991 ; Koopsmans et Boomsma, 1996 ; Maes et coll., 1999 ; Viken et coll., 1999 ; Rose et coll., 2001 ; Hopfer et coll., 2003). De plus, la rapidité dans la transition du premier usage à une consommation régulière est également largement influencée par des facteurs génétiques (Stallings et coll., 1999).Épigénétique et addiction
Le phénotype est le fruit d’interactions continues entre les gènes et l’environnement. Certains signaux intracellulaires régulés par l’environnement, comme les facteurs de transcription, contrôlent l’expression génique. Les interactions physiques entre ces facteurs de transcription et les séquences d’ADN sont trop dynamiques pour expliquer comment les signaux extracellulaires sont capables d’induire des modifications à long-terme de l’expression génique et des fonctions cellulaires. Ceci est particulièrement important au niveau cérébral, où les modifications fonctionnelles persistantes des neurones sont essentielles à certaines fonctions comme la mémoire. La faculté d’organiser de telles adaptations à l’environnement dépend de la capacité des neurones à adapter de façon dynamique la structure et les fonctions génomiques (Meaney et Ferguson-Smith, 2010). Les mécanismes épigénétiques induisent des modifications stables (et pour certaines héritables) de l’expression des gènes ne résultant pas de modifications de la séquence de l’ADN (Jaenisch et Bird, 2003). L’épigénétique englobe divers mécanismes comme la méthylation de l’ADN ou les modifications des histones par acétylation, méthylation ou phosphorylation. Certains de ces mécanismes épigénétiques, comme la méthylation de l’ADN par les DNA méthyltransférases (DNMT), induisent des modifications très stables, tandis que d’autres modifications, comme l’acétylation des histones par les histones acétyltransférases (HAT), seraient plus labiles et moduleraient l’expression des gènes sur des périodes plus courtes (Borrelli et coll., 2008). Ces modifications ou marques épigénétiques modulent la transcription génique et sont essentielles dans le phénomène de plasticité neuronale (Weaver et coll., 2007 ; Borrelli et coll., 2008). Ainsi, les mécanismes épigénétiques permettent d’expliquer les changements d’activité transcriptionnelle associés à la différenciation cellulaire, l’apprentissage et la mémoire, la neurodégénérescence liée à l’âge, le stress chronique, le statut nutritionnel (notamment l’apport en certains micronutriments…) et l’exposition répétée aux toxiques environnementaux et aux drogues (Weaver et coll., 2004 ; Jirtle et Skinner, 2007 ; Renthal et Nestler, 2008 ; Murgatroyd et coll., 2009 ; Roth et Sweatt, 2009).
). Les mécanismes épigénétiques induisent des modifications stables (et pour certaines héritables) de l’expression des gènes ne résultant pas de modifications de la séquence de l’ADN (Jaenisch et Bird, 2003). L’épigénétique englobe divers mécanismes comme la méthylation de l’ADN ou les modifications des histones par acétylation, méthylation ou phosphorylation. Certains de ces mécanismes épigénétiques, comme la méthylation de l’ADN par les DNA méthyltransférases (DNMT), induisent des modifications très stables, tandis que d’autres modifications, comme l’acétylation des histones par les histones acétyltransférases (HAT), seraient plus labiles et moduleraient l’expression des gènes sur des périodes plus courtes (Borrelli et coll., 2008). Ces modifications ou marques épigénétiques modulent la transcription génique et sont essentielles dans le phénomène de plasticité neuronale (Weaver et coll., 2007 ; Borrelli et coll., 2008). Ainsi, les mécanismes épigénétiques permettent d’expliquer les changements d’activité transcriptionnelle associés à la différenciation cellulaire, l’apprentissage et la mémoire, la neurodégénérescence liée à l’âge, le stress chronique, le statut nutritionnel (notamment l’apport en certains micronutriments…) et l’exposition répétée aux toxiques environnementaux et aux drogues (Weaver et coll., 2004 ; Jirtle et Skinner, 2007 ; Renthal et Nestler, 2008 ; Murgatroyd et coll., 2009 ; Roth et Sweatt, 2009).Il est démontré que l’exposition aiguë ou chronique à une drogue induit des modifications de l’expression génique (Yuferov et coll., 2005 ; Bell et coll., 2009 ; Renthal et Nestler, 2009). Cependant, les mécanismes par lesquels les voies de signalisation activées par les drogues aboutissent à ces modifications transcriptionnelles sont encore mal connus. Il s’avère que certains de ces mécanismes sont épigénétiques et participent au remodelage de la chromatine (principalement par acétylation, méthylation ou phosphorylation des histones) ou à la modification de l’ADN (méthylation directe de l’ADN). En effet, des études récentes ont mis en évidence que l’administration de drogue produit des adaptations d’ordre épigénétique chez l’animal (Kim et Shukla, 2006 ; Numachi et coll., 2007 ; Launay et coll., 2009 ; Martin et coll., 2012) comme chez l’Homme (Bonsch et coll., 2004 ; Philibert et coll., 2008 ; Manzardo et coll., 2012). Ces altérations épigénétiques induites par les drogues contribuent directement à des modifications comportementales, comme par exemple la sensibilité aux effets récompensants, aux effets stimulants (Levine et coll., 2005 ; Renthal et coll., 2007 ; Schroeder et coll., 2008 ; Sun et coll., 2008 ; Anier et coll., 2010 ; Malvaez et coll., 2010 ; Legastelois et coll., 2013). Ces découvertes ont ouvert un nouveau champ d’investigation dans le domaine de l’addiction et ont fourni un aperçu des actions des drogues au niveau du nucléosome4
en relation avec l’expression génique et les conséquences physiopathologiques. À l’heure actuelle, parmi tous les mécanismes épigénétiques étudiés dans l’addiction, l’acétylation/désacétylation des histones fait l’objet du plus grand nombre d’études. Ces travaux ont ainsi mis en évidence un rôle crucial de cette balance de régulation dans divers modèles animaux d’addiction (Renthal et Nestler, 2008 ; Robison et Nestler, 2011 ; Wong et coll., 2011).
; Bell et coll., 2009 ; Renthal et Nestler, 2009). Cependant, les mécanismes par lesquels les voies de signalisation activées par les drogues aboutissent à ces modifications transcriptionnelles sont encore mal connus. Il s’avère que certains de ces mécanismes sont épigénétiques et participent au remodelage de la chromatine (principalement par acétylation, méthylation ou phosphorylation des histones) ou à la modification de l’ADN (méthylation directe de l’ADN). En effet, des études récentes ont mis en évidence que l’administration de drogue produit des adaptations d’ordre épigénétique chez l’animal (Kim et Shukla, 2006 ; Numachi et coll., 2007 ; Launay et coll., 2009 ; Martin et coll., 2012) comme chez l’Homme (Bonsch et coll., 2004 ; Philibert et coll., 2008 ; Manzardo et coll., 2012). Ces altérations épigénétiques induites par les drogues contribuent directement à des modifications comportementales, comme par exemple la sensibilité aux effets récompensants, aux effets stimulants (Levine et coll., 2005 ; Renthal et coll., 2007 ; Schroeder et coll., 2008 ; Sun et coll., 2008 ; Anier et coll., 2010 ; Malvaez et coll., 2010 ; Legastelois et coll., 2013). Ces découvertes ont ouvert un nouveau champ d’investigation dans le domaine de l’addiction et ont fourni un aperçu des actions des drogues au niveau du nucléosome4
en relation avec l’expression génique et les conséquences physiopathologiques. À l’heure actuelle, parmi tous les mécanismes épigénétiques étudiés dans l’addiction, l’acétylation/désacétylation des histones fait l’objet du plus grand nombre d’études. Ces travaux ont ainsi mis en évidence un rôle crucial de cette balance de régulation dans divers modèles animaux d’addiction (Renthal et Nestler, 2008 ; Robison et Nestler, 2011 ; Wong et coll., 2011).Ainsi, il a été démontré que les drogues perturbent l’activité des enzymes impliquées dans le remodelage de la chromatine (Botia et coll., 2012). L’alcool par exemple, inhibe l’activité des histones désacétylases. Lorsque les histones sont davantage acétylées, la structure de l’ADN est moins condensée, ce qui la rend plus propice à la transcription génique. Ces mécanismes épigénétiques interviennent aussi certainement lors des expositions aux drogues à une période du développement cérébral comme les expositions in utero ou encore à l’adolescence. Ils pourraient expliquer pourquoi des facteurs environnementaux comme le stress ou la consommation de drogues laissent des « traces » (épigénétiques) à long terme pouvant avoir des répercussions sur le fonctionnement cérébral et le comportement. Des études soulignent la possibilité de cibler ces mécanismes épigénétiques pour réduire, inverser, voire même prévenir la survenue d’une addiction (Naassila, 2012 ; Legastelois et coll., 2013). Ainsi, des travaux ont montré que l’enrichissement social et psychomoteur des individus peut inverser voire prévenir les comportements addictifs, en lien avec la mise en jeu de mécanismes épigénétiques (Solinas et coll., 2010).
). L’alcool par exemple, inhibe l’activité des histones désacétylases. Lorsque les histones sont davantage acétylées, la structure de l’ADN est moins condensée, ce qui la rend plus propice à la transcription génique. Ces mécanismes épigénétiques interviennent aussi certainement lors des expositions aux drogues à une période du développement cérébral comme les expositions in utero ou encore à l’adolescence. Ils pourraient expliquer pourquoi des facteurs environnementaux comme le stress ou la consommation de drogues laissent des « traces » (épigénétiques) à long terme pouvant avoir des répercussions sur le fonctionnement cérébral et le comportement. Des études soulignent la possibilité de cibler ces mécanismes épigénétiques pour réduire, inverser, voire même prévenir la survenue d’une addiction (Naassila, 2012 ; Legastelois et coll., 2013). Ainsi, des travaux ont montré que l’enrichissement social et psychomoteur des individus peut inverser voire prévenir les comportements addictifs, en lien avec la mise en jeu de mécanismes épigénétiques (Solinas et coll., 2010).
En conclusion, depuis trois décennies, les recherches sur la neurobiologie de l’addiction ont permis d’identifier les circuits cérébraux, les systèmes de neurotransmission, ainsi que les dysfonctionnements impliqués dans cette pathologie comportementale complexe, tant sur la vulnérabilité à développer une addiction que sur la susceptibilité à la rechute. Les recherches ont également identifié la période de l’adolescence comme une phase très critique du développement cérébral qui constitue une période de vulnérabilité particulière relativement non seulement aux effets des drogues mais aussi au risque d’abus voire de dépendance. Les enjeux futurs sont : i) l’identification des mécanismes impliqués dans cette vulnérabilité neurodéveloppementale aux drogues (génétiques, épigénétiques, environnementaux) ; ii) une meilleure compréhension des mécanismes impliqués dans la transition de la consommation contrôlée à l’addiction ; iii) l’identification des mécanismes qui jouent un rôle dans les phénomènes de rechute même après une abstinence prolongée ; iv) l’étude de la réactivité des systèmes de neurotransmission suite à la réexposition à la drogue chez les sujets qui ont réussi à s’abstenir ou à retrouver le contrôle de leur consommation. Si les mécanismes qui sous-tendent le passage du plaisir à la dépendance commencent à être bien appréhendés, il n’en est pas de même pour ceux qui peuvent sous-tendre le plaisir de l’abstinence.
Relativement à la problématique de la consommation de drogues chez les jeunes et à la prévention, il est clair que la période de l’adolescence est une période développementale particulièrement critique en termes de vulnérabilité aux effets neurotoxiques et addictifs, non seulement sur le court terme mais aussi à long terme. Il est important de s’attaquer au problème selon les différents angles que sont la dangerosité de l’usage et de la consommation excessive, les représentations sur les produits et leur usage, et le respect de la loi d’interdiction de vente aux mineurs, afin de retarder au maximum l’âge d’initiation et d’entrée dans des consommations régulières ou excessives.
Bibliographie
[1] aarts e, roelofs a, franke b, rijpkema m, fernandez g, et coll. Striatal dopamine mediates the interface between motivational and cognitive control in humans: evidence from genetic imaging.
Neuropsychopharmacology. 2010;
35:1943- 1951
[2] agrawal a, verweij kj, gillespie na, heath ac, lessov-schlaggar cn, et coll. The genetics of addiction-a translational perspective.
Transl Psychiatry. 2012;
17:e140
[3] andersen sl, teicher mh. Delayed effects of early stress on hippocampal development.
Neuropsychopharmacology. 2004;
29:1988- 1993
[4] andersen sl, thompson at, rutstein m, hostetter jc, teicher mh. Dopamine receptor pruning in prefrontal cortex during the periadolescent period in rats.
Synapse. 2000;
37:167- 169
[5] andrucci gl, archer rp, pancoast dl, gordon ra. The relationship of MMPI and Sensation Seeking Scales to adolescent drug use.
J Pers Assess. 1989;
53:253- 266
[6] anier k, malinovskaja k, aonurm-helm a, zharkovsky a, kalda a. DNA methylation regulates cocaine-induced behavioral sensitization in mice.
Neuropsychopharmacology. 2010;
35:2450- 2461
[7] baler rd, volkow nd. Drug addiction: the neurobiology of disrupted self-control.
Trends Mol Med. 2006;
12:559- 566
[8] barnes ca, mcnaughton bl. An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses.
Behav Neurosci. 1985;
99:1040- 1048
[9] batel p, houchi h, daoust m, ramoz n, naassila m, et coll. A haplotype of the DRD1 gene is associated with alcohol dependence.
Alcohol Clin Exp Res. 2008;
32:567- 572
[10] baumrind d. A developmental perspective on adolescent risk taking in contemporary America.
In: Irwin C.E. Jr, editors.
Adolescent Social Behavior and Health.
San Francisco, CA:Jossey-Bass;
1987.
p. 93- 125
[11] bell rl, kimpel mw, mcclintick jn, strother wn, carr lg, et coll. Gene expression changes in the nucleus accumbens of alcohol-preferring rats following chronic ethanol consumption.
Pharmacol Biochem Behav. 2009;
94:131- 147
[12] bell rl, rodd za, lumeng l, murphy jm, mcbride wj. The alcohol-preferring P rat and animal models of excessive alcohol drinking.
Addict Biol. 2006;
11:270- 288
[13] bierut lj, dinwiddie sh, begleiter h, crowe rr, hesselbrock v, et coll. Familial transmission of substance dependence: alcohol, marijuana, cocaine, and habitual smoking: a report from the Collaborative Study on the Genetics of Alcoholism.
Arch Gen Psychiatry. 1998;
55:982- 988
[14] blakemore sj, choudhury s. Development of the adolescent brain: implications for executive function and social cognition.
J Child Psychol Psychiatry. 2006;
47:296- 312
[15] blum k, noble ep, sheridan pj, montgomery a, ritchie t, et coll. Genetic predisposition in alcoholism: association of the D2 dopamine receptor TaqI B1 RFLP with severe alcoholics.
Alcohol. 1993;
10:59- 67
[16] bonsch d, reulbach u, bayerlein k, hillemacher t, kornhuber j, et coll. Elevated alpha synuclein mRNA levels are associated with craving in patients with alcoholism.
Biol Psychiatry. 2004;
56:984- 986
[17] borrelli e, nestler ej, allis cd, sassone-corsi p. Decoding the epigenetic language of neuronal plasticity.
Neuron. 2008;
60:961- 974
[18] botia b, legastelois r, alaux-cantin s, naassila m. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice.
PLoS One. 2012;
7:e47527
[19] casey bj, trainor rj, orendi jl, schubert ab, nystrom le, et coll. A Developmental Functional MRI Study of Prefrontal Activation during Performance of a Go-No-Go Task.
J Cogn Neurosci. 1997;
9:835- 847
[20] cloninger cr, bohman m, sigvardsson s. Inheritance of alcohol abuse. Cross-fostering analysis of adopted men.
Arch Gen Psychiatry. 1981;
38:861- 868
[21] edenberg hj, dick dm, xuei x, tian h, almasy l, et coll. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations.
Am J Hum Genet. 2004;
74:705- 714
[22] ernst m, romeo rd, andersen sl. Neurobiology of the development of motivated behaviors in adolescence: a window into a neural systems model.
Pharmacol Biochem Behav. 2009;
93:199- 211
[23] faden vb. Trends in initiation of alcohol use in the United States 1975 to 2003.
Alcohol Clin Exp Res. 2006;
30:1011- 1022
[24] forbes ee, ryan nd, phillips ml, manuck sb, worthman cm, et coll. Healthy adolescents’ neural response to reward: associations with puberty, positive affect, and depressive symptoms.
J Am Acad Child Adolesc Psychiatry. 2010;
49:162- 172
[25] galvan a, hare ta, parra ce, penn j, voss h, et coll. Earlier development of the accumbens relative to orbitofrontal cortex might underlie risk-taking behavior in adolescents.
J Neurosci. 2006;
26:6885- 6892
[26] galvan a. Adolescent development of the reward system.
Front Hum Neurosci. 2010;
4:6
[27] galvan a, hare t, voss h, glover g, casey bj. Risk-taking and the adolescent brain: who is at risk?.
Dev Sci. 2007;
10:F8- F14
[28] geier cf, terwilliger r, teslovich t, velanova k, luna b. Immaturities in reward processing and its influence on inhibitory control in adolescence.
Cereb Cortex. 2010;
20:1613- 1629
[29] giedd jn. Structural magnetic resonance imaging of the adolescent brain.
Ann N Y Acad Sci. 2004;
1021:77- 85
[30] giedd jn, blumenthal j, jeffries no, castellanos fx, liu h, et coll. Brain development during childhood and adolescence: a longitudinal MRI study.
Nat Neurosci. 1999;
2:861- 863
[31] goldman d, oroszi g, ducci f. The genetics of addictions: uncovering the genes.
Nat Rev Genet. 2005;
6:521- 532
[32] goodwin dw, schulsinger f, moller n, hermansen l, winokur g, guze sb. Drinking problems in adopted and nonadopted sons of alcoholics.
Arch Gen Psychiatry. 1974;
31:164- 169
[33] gorwood p, le sy, ramoz n, dubertret c, moalic jm, et coll. Genetics of dopamine receptors and drug addiction.
Hum Genet. 2012;
131:803- 822
[34] heath ac, martin ng. Teenage alcohol use in the Australian twin register: genetic and social determinants of starting to drink.
Alcohol Clin Exp Res. 1988;
12:735- 741
[35] heath ac, meyer j, jardine r, martin ng. The inheritance of alcohol consumption patterns in a general population twin sample: II. Determinants of consumption frequency and quantity consumed.
J Stud Alcohol. 1991;
52:425- 433
[36] hebb do. The organization of behavior: a neuropsychological theory.
Wiley, New York. 1949;
[37] herman ai, philbeck jw, vasilopoulos nl, depetrillo pb. Serotonin transporter promoter polymorphism and differences in alcohol consumption behaviour in a college student population.
Alcohol Alcohol. 2003;
38:446- 449
[38] hopfer cj, crowley tj, hewitt jk. Review of twin and adoption studies of adolescent substance use.
J Am Acad Child Adolesc Psychiatry. 2003;
42:710- 719
[39] huttenlocher pr. Synapse elimination and plasticity in developing human cerebral cortex.
Am J Ment Defic. 1984;
88:488- 496
[40] hyman se, malenka rc. Addiction and the brain: the neurobiology of compulsion and its persistence.
Nat Rev Neurosci. 2001;
2:695- 703
[41] jaenisch r, bird a. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals.
Nat Genet. 2003;
33 (Suppl):245- 254
[42] jirtle rl, skinner mk. Environmental epigenomics and disease susceptibility.
Nat Rev Genet. 2007;
8:253- 262
[43] kalivas pw, volkow nd. The neural basis of addiction: a pathology of motivation and choice.
Am J Psychiatry. 2005;
162:1403- 1413
[44] kalsbeek a, voorn p, buijs rm, pool cw, uylings hb. Development of the dopaminergic innervation in the prefrontal cortex of the rat.
J Comp Neurol. 1988;
269:58- 72
[45] kaprio j, koskenvuo m, langinvainio h, romanov k, sarna s, rose rj. Genetic influences on use and abuse of alcohol: a study of 5638 adult Finnish twin brothers.
Alcohol Clin Exp Res. 1987;
11:349- 356
[46] kareken da, liang t, wetherill l, dzemidzic m, bragulat v, et coll. A polymorphism in GABRA2 is associated with the medial frontal response to alcohol cues in an fMRI study.
Alcohol Clin Exp Res. 2010;
34:2169- 2178
[47] kasanetz f, deroche-gamonet v, berson n, balado e, lafourcade m, et coll. Transition to addiction is associated with a persistent impairment in synaptic plasticity.
Science. 2010;
328:1709- 1712
[48] kauer ja, malenka rc. Synaptic plasticity and addiction.
Nat Rev Neurosci. 2007;
8:844- 858
[49] kendler ks, schmitt e, aggen sh, prescott ca. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood.
Arch Gen Psychiatry. 2008;
65:674- 682
[50] kim js, shukla sd. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues.
Alcohol Alcohol. 2006;
41:126- 132
[51] koepp mj, gunn rn, lawrence ad, cunningham vj, dagher a, et coll. Evidence for striatal dopamine release during a video game.
Nature. 1998;
393:266- 268
[52] koob gf, le moal m. Review. Neurobiological mechanisms for opponent motivational processes in addiction.
Philos Trans R Soc Lond B Biol Sci. 2008;
363:3113- 3123
[53] koopmans jr, boomsma di. Familial resemblances in alcohol use: genetic or cultural transmission?.
J Stud Alcohol. 1996;
57:19- 28
[54] kostovic i. Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life.
Prog Brain Res. 1990;
85:223- 239
[55] launay jm, del pm, chironi g, callebert j, peoc’h k, et coll. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation.
PLoS One. 2009;
4:e7959
[56] legastelois r, botia b, naassila m. Blockade of ethanol-induced behavioral sensitization by sodium butyrate: descriptive analysis of gene regulations in the striatum.
Alcohol Clin Exp Res. 2013;
37:1143- 1153
[57] levine aa, guan z, barco a, xu s, kandel er, et coll. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum.
Proc Natl Acad Sci U S A. 2005;
102:19186- 19191
[58] li md, burmeister m. New insights into the genetics of addiction.
Nat Rev Genet. 2009;
10:225- 231
[59] limosin f, romo l, batel p, ades j, boni c, et coll. Association between dopamine receptor D3 gene BalI polymorphism and cognitive impulsiveness in alcohol-dependent men.
Eur Psychiatry. 2005;
20:304- 306
[60] lovinger dm, roberto m. Synaptic effects induced by alcohol.
Curr Top Behav Neurosci. 2013;
13:31- 86
[61] maes hh, woodard ce, murrelle l, meyer jm, silberg jl, et coll. Tobacco, alcohol and drug use in eight- to sixteen-year-old twins: the Virginia Twin Study of Adolescent Behavioral Development.
J Stud Alcohol. 1999;
60:293- 305
[62] malvaez m, sanchis-segura c, vo d, lattal km, wood ma. Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference.
Biol Psychiatry. 2010;
67:36- 43
[63] mameli m, luscher c. Synaptic plasticity and addiction: learning mechanisms gone awry.
Neuropharmacology. 2011;
61:1052- 1059
[64] manzardo am, henkhaus rs, butler mg. Global DNA promoter methylation in frontal cortex of alcoholics and controls.
Gene. 2012;
498:5- 12
[65] martin ta, jayanthi s, mccoy mt, brannock c, ladenheim b, et coll. Methamphetamine causes differential alterations in gene expression and patterns of histone acetylation/hypoacetylation in the rat nucleus accumbens.
PLoS One. 2012;
7:e34236
[66] martin ca, kelly th, rayens mk, brogli b, himelreich k, et coll. Sensation seeking and symptoms of disruptive disorder: association with nicotine, alcohol, and marijuana use in early and mid-adolescence.
Psychol Rep. 2004;
94:1075- 1082
[67] meaney mj, ferguson-smith ac. Epigenetic regulation of the neural transcriptome: the meaning of the marks.
Nat Neurosci. 2010;
13:1313- 1318
[68] merikangas kr, risch n. Genomic priorities and public health.
Science. 2003;
302:599- 601
[69] merikangas kr, stevens de, fenton b, stolar m, o’malley s, et coll. Co-morbidity and familial aggregation of alcoholism and anxiety disorders.
Psychol Med. 1998;
28:773- 788
[70] murgatroyd c, patchev av, wu y, micale v, bockmuhl y, et coll. Dynamic DNA methylation programs persistent adverse effects of early-life stress.
Nat Neurosci. 2009;
12:1559- 1566
[71] naassila m. Du plaisir à la dépendance.
Cerveau & Psycho. 2008, N° 29 - septembre-octobre;
[72] naassila m. Play on the epigenetic to counter addiction to alcohol.
Biofutur. Décembre 2012;
338:34- 35
[73] naassila m. Alcohol and rats.
Elsevier Encyclopedia on Addictive behaviors, Biological Research on Addiction, Volume 2. 2013. Biological Research on Addiction;
(Volume 2. Elsevier Inc)
[74] nees f, witt sh, lourdusamy a, vollstadt-klein s, steiner s, et coll. Genetic risk for nicotine dependence in the cholinergic system and activation of the brain reward system in healthy adolescents.
Neuropsychopharmacology. 2013;
38:2081- 2089
[75] numachi y, shen h, yoshida s, fujiyama k, toda s, et coll. Methamphetamine alters expression of DNA methyltransferase 1 mRNA in rat brain.
Neurosci Lett. 2007;
414:213- 217
[76] olds j, milner p. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain.
J Comp Physiol Psychol. 1954;
47:419- 427
[77] olsson ca, byrnes gb, lotfi-miri m, collins v, williamson r, et coll. Association between 5-HTTLPR genotypes and persisting patterns of anxiety and alcohol use: results from a 10-year longitudinal study of adolescent mental health.
Mol Psychiatry. 2005;
10:868- 876
[78] pagan jl, rose rj, viken rj, pulkkinen l, kaprio j, et coll. Genetic and environmental influences on stages of alcohol use across adolescence and into young adulthood.
Behav Genet. 2006;
36:483- 497
[79] philibert ra, sandhu h, hollenbeck n, gunter t, adams w, et coll. The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies.
Am J Med Genet B Neuropsychiatr Genet. 2008;
147B:543- 549
[80] prescott ca, kendler ks. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins.
Am J Psychiatry. 1999;
156:34- 40
[81] prescott ca, cross rj, kuhn jw, horn jl, kendler ks. Is risk for alcoholism mediated by individual differences in drinking motivations?.
Alcohol Clin Exp Res. 2004;
28:29- 39
[82] ramchandani va, umhau j, pavon fj, ruiz-velasco v, margas w, et coll. A genetic determinant of the striatal dopamine response to alcohol in men.
Mol Psychiatry. 2011;
16:809- 817
[83] renthal w, nestler ej. Epigenetic mechanisms in drug addiction.
Trends Mol Med. 2008;
14:341- 50
[84] renthal w, nestler ej. Histone acetylation in drug addiction.
Semin Cell Dev Biol. 2009;
20:387- 394
[85] renthal w, maze i, krishnan v, covington he, iii, xiao g, et coll. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli.
Neuron. 2007;
56:517- 529
[86] rhee sh, hewitt jk, young se, corley rp, crowley tj, et coll. Genetic and environmental influences on substance initiation, use, and problem use in adolescents.
Arch Gen Psychiatry. 2003;
60:1256- 1264
[87] robison aj, nestler ej. Transcriptional and epigenetic mechanisms of addiction.
Nat Rev Neurosci. 2011;
12:623- 637
[88] rose rj, dick dm, viken rj, pulkkinen l, kaprio j. Drinking or abstaining at age 14? A genetic epidemiological study.
Alcohol Clin Exp Res. 2001;
25:1594- 1604
[89] rosenberg dr, lewis da. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study.
Biol Psychiatry. 1994;
36:272- 277
[90] roth tl, sweatt jd. Regulation of chromatin structure in memory formation.
Curr Opin Neurobiol. 2009;
19:336- 342
[91] schroeder fa, penta kl, matevossian a, jones sr, konradi c, et coll. Drug-induced activation of dopamine D(1) receptor signaling and inhibition of class I/II histone deacetylase induce chromatin remodeling in reward circuitry and modulate cocaine-related behaviors.
Neuropsychopharmacology. 2008;
33:2981- 2992
[92] schultz w. Dopamine signals for reward value and risk: basic and recent data.
Behav Brain Funct. 2010;
6:24
[93] schuckit ma. A long-term study of sons of alcoholics.
Alcohol Health & Research World. 1995;
19:172- 175
[94] solinas m, thiriet n, chauvet c, jaber m. Prevention and treatment of drug addiction by environmental enrichment.
Prog Neurobiol. 2010;
92:572- 592
[95] sowell er, thompson pm, holmes cj, jernigan tl, toga aw. In vivo evidence for post-adolescent brain maturation in frontal and striatal regions.
Nat Neurosci. 1999;
2:859- 861
[96] sowell er, thompson pm, tessner kd, toga aw. Mapping continued brain growth and gray matter density reduction in dorsal frontal cortex: Inverse relationships during postadolescent brain maturation.
J Neurosci. 2001;
21:8819- 8829
[97] spear lp. The adolescent brain and age-related behavioral manifestations.
Neurosci Biobehav Rev. 2000;
24:417- 463
[98] stacey d, bilbao a, maroteaux m, jia t, easton ac, et coll. RASGRF2 regulates alcohol-induced reinforcement by influencing mesolimbic dopamine neuron activity and dopamine release.
Proc Natl Acad Sci USA. 2012;
109:21128- 21133
[99] stallings mc, hewitt jk, beresford t, heath ac, eaves lj. A twin study of drinking and smoking onset and latencies from first use to regular use.
Behav Genet. 1999;
29:409- 421
[100] sun j, wang l, jiang b, hui b, lv z, et coll. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats.
Neurosci Lett. 2008;
441:72- 76
[101] tamm l, menon v, reiss al. Maturation of brain function associated with response inhibition.
J Am Acad Child Adolesc Psychiatry. 2002;
41:1231- 1238
[102] treutlein j, kissling c, frank j, wiemann s, dong l, et coll. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples.
Mol Psychiatry. 2006;
11:594- 602
[103] van der zwaluw cs, kuntsche e, engels rc. Risky alcohol use in adolescence: the role of genetics (DRD2, SLC6A4) and coping motives.
Alcohol Clin Exp Res. 2011;
35:756- 764
[104] van eden cg, kros jm, uylings hb. The development of the rat prefrontal cortex. Its size and development of connections with thalamus, spinal cord and other cortical areas.
Prog Brain Res. 1990;
85:169- 183
[105] van leijenhorst l, gunther mb, op de macks za, rombouts sa, westenberg pm, et coll. Adolescent risky decision-making: neurocognitive development of reward and control regions.
Neuroimage. 2010;
51:345- 355
[106] vanderschuren lj, pierce rc. Sensitization processes in drug addiction.
Curr Top Behav Neurosci. 2010;
3:179- 195
[107] vaughn mg, beaver km, delisi m, howard mo, perron be. Dopamine D4 receptor gene exon III polymorphism associated with binge drinking attitudinal phenotype.
Alcohol. 2009;
43:179- 184
[108] verweij kj, zietsch bp, lynskey mt, medland se, neale mc, et coll. Genetic and environmental influences on cannabis use initiation and problematic use: a meta-analysis of twin studies.
Addiction. 2010;
105:417- 430
[109] viken rj, kaprio j, koskenvuo m, rose rj. Longitudinal analyses of the determinants of drinking and of drinking to intoxication in adolescent twins.
Behav Genet. 1999;
29:455- 461
[110] wang jc, kapoor m, goate am. The genetics of substance dependence.
Annu Rev Genomics Hum Genet. 2012;
13:241- 261
[111] weaver ic, cervoni n, champagne fa, d’alessio ac, sharma s, et coll. Epigenetic programming by maternal behavior.
Nat Neurosci. 2004;
7:847- 854
[112] weaver ic, d’alessio ac, brown se, hellstrom ic, dymov s, et coll. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes.
J Neurosci. 2007;
27:1756- 1768
[113] wills ta, vaccaro d, mcnamara g. Novelty seeking, risk taking, and related constructs as predictors of adolescent substance use: an application of Cloninger’s theory.
J Subst Abuse. 1994;
6:1- 20
[114] wong cc, mill j, fernandes c. Drugs and addiction: an introduction to epigenetics.
Addiction. 2011;
106:480- 489
[115] yuferov v, nielsen d, butelman e, kreek mj. Microarray studies of psychostimulant-induced changes in gene expression.
Addict Biol. 2005;
10:101- 118
[116] zecevic n, bourgeois jp, rakic p. Changes in synaptic density in motor cortex of rhesus monkey during fetal and postnatal life.
Brain Res Dev Brain Res. 1989;
50:11- 32
→ Aller vers SYNTHESE