| |
| Med Sci (Paris). 2013 June; 29(6-7): 637–641. Published online 2013 July 12. doi: 10.1051/medsci/2013296017.L’analyse ADN dans l’approche anthropologique des populations du passé Christine Keyser,1 Éric Crubézy,2 and Bertrand Ludes1* 1Institut de médecine légale, laboratoire d’anthropologie moléculaire, CNRS UMR 5288, 11, rue Humann, 67085Strasbourg, France 2Laboratoire d’anthropologie moléculaire et imagerie de synthèse (AMIS), CNRS UMR 5288, 37, allée Jules Guesde, 31000Toulouse, France |
Grâce à l’analyse de l’ADN présent dans les tissus humains anciens et au développement de marqueurs génétiques qui permettent d’explorer de courtes séquences de cette molécule, il est actuellement possible d’étudier les populations du passé, leur structure, leur migration, ainsi que la dynamique de peuplement de certaines régions du monde. En effet, l’étude du profil génétique de sujets inhumés dans des nécropoles permet de déterminer leurs liens de parenté avec d’autres sujets, d’estimer leur origine biogéographique ainsi que les voies de migration qu’ils ont pu emprunter il y a des centaines ou milliers d’années, de comparer leurs caractéristiques génétiques avec celles des populations actuelles, ou encore d’établir certains de leurs traits physiques. Ces études reposent sur une collaboration étroite des « paléogénéticiens » avec les anthropologues présents sur le site de fouilles, notamment pour préciser les conditions taphonomiques1 qui influent sur la conservation de l’ADN et éviter toute contamination par de l’ADN exogène lors du prélèvement [
14]. Cette collaboration doit s’étendre aux biologistes moléculaires, aux biostatisticiens et aux généticiens des populations pour l’interprétation des résultats obtenus qui se réfèrent souvent à un faible nombre d’individus. |
Matériel et techniques d’études Matériel Les analyses d’ADN ancien sont le plus fréquemment réalisées à partir de tissus osseux et dentaires qui possèdent un microenvironnement favorable à la conservation de fragments d’ADN grâce à leur adsorption sur une matrice minérale, l’hydroxyapatite [
1]. Mais des analyses peuvent également être réalisées à partir de tissus mous momifiés ou de restes pileux. L’exploitation de ces tissus dépend de leurs conditions de conservation et de leur degré de contamination par de l’ADN moderne. L’analyse simultanée, quand elle est possible, de plusieurs prélèvements tissulaires différents pour un même individu (os, dent et/ou cheveux) permet d’authentifier les résultats obtenus et donc d’écarter toute contamination par de l’ADN moderne. ADN et marqueurs moléculaires utilisés Les premiers marqueurs développés pour analyser des séquences d’ADN ancien ont ciblé l’ADN mitochondrial (ADNmt). En effet, cet ADN est présent à de multiples exemplaires dans le cytoplasme des cellules à la différence de l’ADN nucléaire [
15]. De plus, il est circulaire et de petite taille et résiste donc mieux aux facteurs de dégradation peut-être en s’adsorbant plus aisément aux molécules d’hydroxyapatite. Toutefois, les régions de variabilité portées par l’ADNmt ne permettent pas toujours de répondre à la problématique posée, car cet ADN ne représente qu’une partie de l’histoire du génome. Une autre limite liée à l’analyse de l’ADNmt est sa transmission qui est inchangée de génération en génération (en raison de l’absence de recombinaison) et qui est exclusivement maternelle. Ainsi, son analyse ne permet de retracer que les lignées maternelles. Le développement de marqueurs génétiques à visée nucléaire a permis, à la suite des précédentes analyses de l’ADNmt, d’analyser l’ADN contenu dans le noyau des cellules. Cet ADN, certes plus sensible aux dégradations, est transmis sur un mode mendélien à l’exception de la partie non recombinante du chromosome Y. L’étude des marqueurs nucléaires autosomaux (chromosomes non sexuels) permet de retracer des liens de proche parenté, tandis que l’analyse spécifique de marqueurs situés sur la partie non recombinante du chromosome Y permet l’étude des lignées paternelles. Alors que le chromosome Y et l’ADNmt sont très utiles pour retracer l’origine biogéographique et les migrations des populations du passé, l’ADN nucléaire permet, outre la détermination des liens de proche parenté, la caractérisation des traits physiques des individus composant ces populations. Deux types de marqueurs moléculaires sont classiquement utilisés pour l’étude de l’ADN conservé dans des tissus anciens : les microsatellites ou STR (short tandem repeats) et les polymorphismes ponctuels de séquences (SNP, single nucleotide polymorphisms). Notons néanmoins que l’avènement des nouvelles générations de séquenceurs (NGS) [
2] permet désormais d’étendre les investigations moléculaires à l’analyse de tous les types de polymorphismes, et surtout à celle de génomes complets [
3,
4, 15,
16]. Les STR sont formés d’unités de deux à sept nucléotides, répétées à un même endroit de l’ADN, en un nombre qui varie beaucoup selon les individus. Ceux présents sur les autosomes permettent d’établir un profil génétique propre à chaque individu et de l’identifier, tandis que ceux localisés sur les chromosomes sexuels permettent de retracer les filiations patrilinéaires à l’aide du chromosome Y et matrilinéaires (en partie seulement) par le chromosome X. L’analyse des STR portés par le chromosome Y définissent un haplotype Y caractéristique d’une lignée paternelle. Les STR sont localisés sur des régions non codantes de l’ADN nucléaire (ne donnant pas d’expression phénotypique), au contraire des mutations ponctuelles de séquence (SNP), présentes aussi bien sur des régions codantes que non codantes de l’ADN nucléaire et de l’ADN mitochondrial. Les SNP mitochondriaux permettent de définir des haplotypes qui caractérisent des lignées maternelles, ainsi que des haplogroupes qui rassemblent les haplotypes ayant une origine ancestrale commune. Les SNP présents sur les autosomes donnent la possibilité de déterminer des caractéristiques phénotypiques, telles que la couleur de la peau, des yeux ou des cheveux. Ceux portés par les chromosomes Y et l’ADN mitochondrial permettent de définir des haplogroupes qui présentent une spécificité géographique. Les STR situés sur les autosomes ou les chromosomes sexuels sont actuellement amplifiés à l’aide de kits produits de façon industrielle (par exemple Applied Biosystems, Promega ou Qiagen) pour l’identification humaine en criminalistique. Ils permettent une amplification simultanée de nombreuses régions STR, réduisant ainsi la quantité d’ADN nécessaire pour une amplification par réaction de polymérisation en chaîne (PCR). Les produits amplifiés sont marqués par différents fluorochromes et peuvent donc être identifiés sur un séquenceur automatique. En ce qui concerne l’ADN mitochondrial, l’une des régions les plus étudiées se situe au niveau de la région de contrôle (D-Loop), une région non codante qui comporte des régions hypervariables. L’analyse est le plus souvent réalisée par séquençage classique, permettant la détermination d’environ 600 à 700 nucléotides. La séquence établie est ensuite comparée à une séquence de référence publiée par Anderson en 1981 [
5,
6]. Mais d’autres polymorphismes sont également recherchés sur la partie codante de la molécule et identifiés au moyen de différentes techniques de typage des SNP, permettant ainsi une détermination plus précise des haplogroupes auxquels rattachés les haplotypes. Mesures prises pour éviter les contaminations En raison des risques importants de contamination des échantillons anciens par de l’ADN contemporain, les restes biologiques anciens sont manipulés avec de grandes précautions (port de gants, de masques faciaux, de charlottes) et traités selon des règles strictes (locaux réservés au traitement de l’ADN ancien, séparation stricte des espaces pré- et post-PCR, etc.). Les résultats obtenus à partir des échantillons anciens sont comparés aux profils génétiques de toutes les personnes ayant été en contact avec ces échantillons lors de la fouille, lors du transport ou au niveau du laboratoire, et les analyses sont répétées au minimum à trois reprises pour attester de la validité des résultats. |
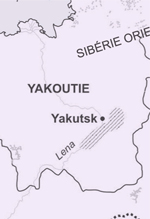
Exemple de l’analyse des populations sibériennes du passé Pour illustrer notre propos, prenons l’exemple de nos travaux sur le peuplement de la Sibérie, qui s’intéressent à deux régions principales : la vallée de l’Ienissei (sud de la Sibérie) et la Yakoutie (Sibérie orientale) (Figure 1).
 | Figure 1.
Localisation géographique des populations sibériennes du passé analysées. Deux échantillons anciens ont été principalement analysés : celui de la vallée de l’Ienissei (Krasnoyarsk, sud de la Sibérie) et celui de la Yakoutie (Yakutsk, Sibérie orientale). |
La vallée de l’Ienissei Les données concernant l’origine et l’histoire des populations qui peuplaient le sud de la Sibérie par le passé sont assez pauvres. Pour cette raison et grâce à une collaboration avec l’université de Krasnoyarsk (Russie), nous avons étudié des sujets de l’âge du bronze et de l’âge du fer exhumés de kourganes 2 localisés dans la région de Krasnoyarsk et appartenant à différentes cultures qui, pour certaines, sont considérées comme indo-européennes. Les kourganes ou tumulus sont des constructions caractéristiques d’une culture apparue dans les steppes de la Russie du Sud aux environs de 5 000 ans avant Jésus-Christ et qui se serait ensuite étendue vers l’est, le centre et le nord de l’Europe entre 4 400 et 2 800 ans avant Jésus-Christ. Nous avons étudié 26 restes osseux humains datés du milieu du iie millénaire avant Jésus-Christ au ive siècle après Jésus-Christ. Les analyses de l’ADN mitochondrial ont montré que ces spécimens anciens étaient porteurs à la fois de lignées européennes et asiatiques. Néanmoins, la plupart des séquences mitochondriales obtenues (77 %) appartenaient à des haplogroupes mitochondriaux européens (HV, H, T, I, U, et K), et seules 23 % correspondaient à des lignées asiatiques (C, Z, G2a, F1b et N9a). La proportion de séquences européennes atteignait 90 % pour l’âge du bronze, ce qui valide l’hypothèse d’une prédominance de séquences européennes aux ie et iie millénaires avant Jésus-Christ. À l’âge du fer, cette prédominance semblait s’estomper au profit de séquences asiatiques, les séquences européennes ne représentant plus que de 67 %. Ce résultat posait la question de l’origine de ces individus porteurs de séquences mitochondriales européennes. D’après la littérature, il semblait que la composante européenne ou caucasoïde observée dans notre échantillon ancien puisse provenir des populations d’Europe de l’Est [
7,
8]. Les analyses génétiques réalisées sur le chromosome Y ont montré que, à une exception près, tous les échantillons masculins (10 sujets/26), bien que sans lien de proche parenté (comme démontré par l’analyse de STR), appartenaient à l’haplogroupe R1a1a. Cet haplogroupe est présent à des fréquences élevées sur tout le continent eurasien. Selon certains auteurs, la distribution géographique de l’haplogroupe R1a1a pourrait être le reflet du repeuplement de l’Europe après la dernière période de glaciation (20 à 12 000 ans) depuis une région refuge située dans l’est de l’Europe, vraisemblablement en Ukraine. Il pourrait, en outre, refléter l’expansion des Indo-Européens [
9]. Les données archéologiques ne permettent pas d’approcher les caractères morphologiques des populations étudiées et donc d’établir la présence de traits de type européen chez les sibériens anciens. Donc, nous avons poursuivi nos investigations en analysant des marqueurs génétiques (SNP) localisés au sein de gènes impliqués dans le mécanisme de la pigmentation. Ces polymorphismes entraînent des variations qui intéressent à la fois la couleur des yeux, des cheveux et de la peau, et qui permettent de déterminer soit un phénotype, soit une origine biogéographique [
10]. À partir des résultats obtenus pour les 26 Sibériens anciens étudiés, nous avons déterminé que près de 60 % d’entre eux avaient les yeux clairs et que la plupart devaient avoir les cheveux et la peau claire. Pour ce qui est de leur origine biogéographique, 23 semblaient d’origine européenne, deux d’origine asiatique et une d’origine mixte [
11]. Notre étude a montré que des individus de la culture dite des Kourganes étaient porteurs de l’haplogroupe R1a1a, haplogroupe qui pourrait être l’un des marqueurs de migration des premiers indo-européens. Ainsi, l’histoire du peuplement du sud sibérien pourrait être liée à l’expansion des Indo-Européens. Elle a également démontré pour la 1e fois que des individus issus de populations du sud sibérien datées de l’âge du bronze et de l’âge du fer possédaient des caractères physiques de type européen. D’où venaient précisément ces individus ? Voilà l’une des questions auxquelles nous tentons de répondre par l’analyse de nouveaux spécimens. La Yakoutie L’ethnogenèse du peuple yakoute est peu connue, mais il semble que ce groupe ethnique soit le résultat d’une migration vers le nord de populations mongoles et/ou turques venues d’Asie centrale ou du sud de la Sibérie, il y a 800 à 1 200 ans environ. En effet, les Yakoutes sont des pasteurs semi-nomades pratiquant l’élevage de bovidés et de chevaux et parlant une langue appartenant à la famille des langues turques. Il s’agit d’une population isolée d’un point de vue linguistique et dans leur mode de vie par rapport aux populations voisines, dont les Evenks et les Evens qui parlent une langue Tongousse et sont traditionnellement des chasseurs/cueilleurs et des éleveurs de rennes. Les Yakoutes semblent donc davantage liés aux populations de la steppe qu’aux populations sibériennes. Toutefois, la colonisation russe dès 1629 a modifié la composition des différentes ethnies de Sibérie. Afin de comprendre la dynamique de peuplement de la Yakoutie, nous avons analysé les prélèvements réalisés sur des individus inhumés en Yakoutie entre le xve et le xviiie siècle, soit avant la colonisation russe. Ces prélèvements ont été collectés lors de fouilles menées en Yakoutie Centrale, à Verkoïansk et en Villuy par l’équipe de la MAFSO (Mission archéologique française en Sibérie orientale). Ce travail, qui s’est étendu sur 10 ans, a permis l’analyse de plus de 130 individus yakoutes anciens, tant au niveau de l’ADN nucléaire que de l’ADN mitochondrial. Nous avons tout d’abord analysé au moyen de STR autosomaux les relations de proche parenté qui peuvent exister entre les individus inhumés sur un même site funéraire, et nous avons observé des parentés biologiques entre sujets exhumés de sépultures multiples. Nous avons notamment pu établir que l’une de ces tombes contenait une mère, ses deux enfants et deux de ses petits-enfants. L’analyse du chromosome Y, quant à elle, a révélé une faible diversité haplotypique des lignées paternelles, vraisemblablement issue d’un nombre réduit d’individus. La comparaison avec les populations actuelles a mis en évidence que les lignées majoritaires sont propres aux populations yakoutes anciennes et actuelles. Combinés aux données sur l’ADN mitochondrial, ces résultats ont permis de formuler l’hypothèse selon laquelle la colonisation de la Yakoutie est le résultat d’un effet fondateur : un petit nombre de sujets, originaires de la région autour du lac Baïkal, auraient migré vers le nord en suivant, très vraisemblablement, le fleuve la Lena (Figure 1). Ce groupe se serait ensuite mélangé aux autochtones, et notamment aux Evenks [
12]. Pour autant, ce schéma de migration mérite d’être affiné et les investigations se poursuivent actuellement. |
L’analyse combinée de marqueurs à transmission uniparentale (portés par l’ADN mitochondrial et le chromosome Y) et biparentale (présents sur les autosomes) permet d’apporter des éléments cruciaux pour retracer l’histoire des populations humaines du passé ou imaginer à quoi pouvait ressembler nos ancêtres. Les progrès prodigieux accomplis dans les méthodes de séquençage nous permettront d’aller bien au-delà dans un temps très court. En effet, l’analyse du génome complet de deux hominidés anciens (hommes de Néandertal et de Denisova) a d’ores et déjà permis d’obtenir des informations sur les flux de gènes entre groupes humains archaïques, leurs dates de divergence, leurs traits phénotypiques ou leurs différences biologiques [3, 4]. Nul doute que les études de paléogénomique, appliquées dans un futur proche aux populations de cimetières, nous livreront un nombre impressionnant de données sur notre passé [
13]. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
|
Footnotes |
1.
Brundin
M
,
Figdor
D
,
Sundgvist
G
,
Sjögren
U
. DNA binding to hydroxyapatite: a potential mechanism for preservation of microbial DNA . J Endod.
2013; ; 39 : :211.–216. 2.
Stoppa-Lyonnet
D
, Houdayer. Séquençage de nouvelle génération en génétique médicale : du génotype au phénotype, un défi majeur . Med Sci (Paris).
2012; ; 28 : :123.–124. 3.
Green
RE
,
Krause
J
,
Briggs
AW
, et al.
A draft sequence of the Neandertal genome . Science.
2010; ; 328 : :710.–722. 4.
Reich
D
,
Green
RE
,
Kircher
M
, et al.
Genetic history of an archaic hominin group from Denisova Cave in Siberia . Nature.
2010; ; 468 : :1053.–1060. 5.
Anderson
S
,
Bankier
AT
,
Barrell
BG
, et al.
Sequence and organisation of the human mitochondrial genome . Nature.
1981; ; 290 : :457.–446. 6.
Andrews
RM
,
Kubacka
I
,
Chinnery
PF
, et al.
Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA . Nat Genet . , 1999; ; 23 : :147.. 7.
Rosser
ZH
,
Zerjal
T
,
Hurles
ME
, et al.
Y-chromosomal diversity in Europe is clinal and influenced primarily by geography rather than by language . Am J Hum Genet.
2000; ; 67 : :1526.–1543. 8.
Semino
O
,
Passarino
G
,
Oefner
PJ
, et al.
The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective . Science.
2000; ; 290 : :1155.–1159. 9.
Passarino
G
,
Semino
O
,
Magri
C
, et al.
The 49a, f haplotype 11 is a new marker of the EU19 lineage that traces migrations from northern regions of the Black Sea . Hum Immunol.
2001; ; 62 : :922.–932. 10.
Bouakaze
C
,
Keyser
C
,
Crubézy
E
, et al.
Pigment phenotype and biogeographical ancestry from ancient skeletal remains: inferences from multiplexed autosomal SNP analysis . Int J Legal Med.
2009; ; 129 : :395.–410. 11.
Keyser
C
,
Bouakaze
C
,
Crubézy
E
, et al.
Ancient DNA provides new insights into the history of south Siberian Kurgan people . Hum Genet.
2009; ; 126 : :395.–410. 12.
Crubézy
E
,
Amory
S
,
Keyser
C
, et al.
Human evolution in Siberia: from frozen bodies to ancient DNA . BMC Evol Biol.
2010; ; 10 : :25.. 13.
Froment
A
. Génétique et évolution de l’homme en Afrique . Med Sci (Paris).
2010; ; 26 : :17.–18. 14.
Debruyne
R
,
Barriel
V
. Évolution biologique et ADN ancien . Med Sci (Paris).
2006; ; 22 : :502.–506. 15.
Gilgenkrantz
S
. Les prémices du génome de Néandertal . Med Sci (Paris).
2007; ; 23 : :95.–98. 16.
Jordan
B
. Néandertal et Homo sapiens : To meet, or not to meet ?
Med Sci (Paris).
2012; ; 28 : :1129.–1132. |


