| |
| Med Sci (Paris). 2013 November; 29(11): 991–997. Published online 2013 November 20. doi: 10.1051/medsci/20132911015.Oncogène Kras et cancer du pancréas Trente ans plus tard Barbara Bournet,1,2 Marlène Dufresne,1 Janick Selves,1 Jérôme Torrisani,1 Pierre Cordelier,1 and Louis Buscail1,2* 1Inserm UMR U1037, Centre de recherche sur le cancer de Toulouse, CHU Rangueil, 1, avenue Jean Poulhès, BP 84225, 31432Toulouse Cedex 4, France 2Service de gastroentérologie et nutrition, CHU Rangueil-Larrey, 1, avenue Jean Poulhès, TSA 50032, 31059Toulouse, Cedex 9, France |
Les oncogènes proviennent de gènes cellulaires normaux (proto-oncogènes) contrôlant les étapes de la croissance et de la différenciation cellulaires. Lorsqu’ils sont activés (surexpression, mutation ponctuelle), ils contribuent à la transformation maligne de la cellule. D’une façon générale, une mutation ponctuelle de l’un ou des deux allèles d’un oncogène se traduit par une dérégulation de la fonction de la protéine qu’il code. Les oncogènes sont classés selon leurs propriétés en familles : facteurs de croissance, récepteurs de facteurs de croissance, protéines impliquées dans la transduction des signaux prolifératifs, et protéines nucléaires régulant le cycle cellulaire. Les gènes de la famille Ras, Hras, Nras et Kras (H pour Harvey, N pour neuroblastoma, et K pour Kirsten), codent pour une protéine de 21 kDa impliquée dans la transduction des signaux prolifératifs des facteurs de croissance. Les protéines p21-Ras assurent leur fonction de transduction en se liant au GTP. La régulation fine du degré d’activation de p21-Ras se trouve déstabilisée lorsqu’il existe une mutation au niveau du site de liaison du GTP. La conséquence fonctionnelle de cette mutation est une suractivation des signaux prolifératifs transmise en cascade aux protéines de la transduction situées en aval de p21-Ras (sérine et thréonine kinases). Parmi les cancers épithéliaux, l’adénocarcinome pancréatique est celui qui présente le plus fréquemment une mutation de l’oncogène Kras, participant au gain de prolifération et d’invasion pour ce cancer de mauvais pronostic et difficile à traiter [
1,
2]. Plus de 30 ans après la découverte de Kras, le but de cette revue est de rapporter l’intérêt de l’étude de cet oncogène dans le cancer du pancréas, tant au plan scientifique que médical. Nous aborderons plus particulièrement la pathologie expérimentale et l’implication possible dans le diagnostic du cancer du pancréas. |
Rôle et conséquences de l’activation du gène Ras Le gène Kras se situe sur le chromosome 12 (12pl2). La protéine p21-Ras est cytosolique et son ancrage à la face interne de la membrane cytoplasmique par son extrémité carboxy-terminale est nécessaire pour son activation [2]. Elle présente des homologies de structure avec la sous-unité α des protéines G. L’activation de la protéine Ras se fait en plusieurs étapes successives au cours desquelles Ras subira des modifications post-traductionnelles au niveau du domaine CAAX de sa région carboxy-terminale (pour une revue voir [
33]). Dans un premier temps, la protéine Ras nouvellement synthétisée va subir une étape de farnésylation (par une farnésyltransférase cytosolique) au niveau de la cystéine du domaine CAAX, augmentant ainsi l’hydrophobicité de cette région et promouvant l’association de Ras à la membrane du réticulum endoplasmique. À ce site réticulaire, le clivage de CAAX (par la Ras-converting enzyme 1) et la carboxyméthylation des résidus farnésylcystéine amino-terminaux (par une isoprenylcysteine carboxymethyltransferase) génère une forme de Ras qui s’associe préférentiellement sur la face cytosolique des membranes de l’appareil de Golgi. Une dernière étape de palmytoylation de Ras (par une palmitoyltransférase golgienne) va permettre son association à des vésicules intracellulaires et son adressage final à la membrane plasmique. La deuxième étape comporte les échanges GDP/GTP (guanosine di- et triphosphate) : les protéines Ras ont une activité GTPase intrinsèque faible et fonctionnent comme un interrupteur GDP/GTP. La forme liée au GTP est active, tandis que celle liée au GDP ne l’est pas. Les facteurs d’échanges de nucléotides guanidiques permettent l’échange de GDP en GTP lors de l’activation de Ras. De leur côté, les GTPase-activating proteins stimulent l’activité GTPasique de p21-Ras, transformant le GTP en GDP aboutissant ainsi à l’inactivation de la protéine. P21-Ras transmet les signaux prolifératifs des facteurs de croissance se liant aux récepteurs à sept domaines transmembranaires couplés aux protéines G (RCPG)1 et aux récepteurs à activité tyrosine kinase comme l’EGFR (epidermal growth factor receptor). Après activation de p21-Ras, un certain nombre de signaux de transduction vont être activés en cascade. Les protéines Raf (A-Raf, B-Raf et Raf-1) sont d’abord recrutées par p21-Ras et vont activer les kinases MAPK/MEK1 et -2 (mitogen-activated extracellular signal regulated kinases) [
31]. MEK1 et MEK2 ont une double activité tyrosine et sérine/thréonine kinase, et elles activent par phosphorylation les kinases ERK1 et -2 (extracellular signal-regulated serine/threonine kinase 1/2). Elles vont activer à leur tour par phosphorylation leurs cibles nucléaires ou cytoplasmiques au rang desquelles des kinases effectrices comme p90RSK (p90 ribosomal S6 kinase) et des facteurs de transcription (dont c-Fos, c-Jun et c-Myc) (Figure 1) [1–
3]. Ces facteurs de transcription sont impliqués dans le contrôle de la prolifération et du cycle cellulaires (via p16, p15, p21, p27 et la cycline D1), de la différenciation et de la survie cellulaires (via p90RSK), de la transition épithéliomésenchymateuse, de l’invasion et de la migration cellulaires (via le TGFβ [transforming growth factor β]), et enfin de l’angiogenèse (via Raf et VEGF [vascular endothelial growth factor]). La PI3K (phosphoinositide 3-kinase) est aussi un des effecteurs de la voie Ras conduisant à la phosphorylation du PIP2, puis à l’activation de PIP3 et d’AKT (protéine kinase B). Cette dernière va jouer un rôle dans la survie cellulaire et le contrôle de l’apoptose.
 | Figure 1.
Mécanismes moléculaires mis en jeu par la mutation de l’oncogène Kras. |
La mutation du gène Kras modifie le site de liaison du GTP dans la protéine p21-Ras. L’activité GTPasique de p21-Ras mutée est réduite, ainsi que sa capacité à interagir avec les GTPase activating proteins. La conséquence fonctionnelle de cette mutation est une suractivation, puisque p21-Ras est en permanence liée au GTP. Les signaux mitogènes, de migration, d’invasion, d’angiogenèse sont donc activés en permanence sans activation préalable induite par la fixation d’un facteur de croissance ou pro-angiogénique à son récepteur. |
Mutation de l’oncogène Kras et carcinogenèse pancréatique La mutation ponctuelle de l’oncogène Kras confère donc un gain de prolifération non contrôlée à la cellule cancéreuse pancréatique qui acquiert ainsi un pouvoir oncogénique amplifié. Cette mutation siège au niveau des exons 1 et 2 et, plus particulièrement, au niveau du codon 12 (mutation de loin la plus fréquente au niveau de la première ou deuxième base), et plus rarement au niveau des codons 13, 19, 59 et 61 [2]. La forme sauvage du codon 12 est GGT (glycine) ; les formes mutées les plus fréquentes sont GAT (acide aspartique) et GTT (valine). Pour le codon 13, une glycine est transformée le plus souvent en acide aspartique (GAT) (Figure 1) [1,
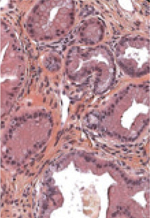
4]. Cette mutation est retrouvée dans 75 à 95 % des pièces d’exérèse d’adénocarcinome pancréatique. Elle est également présente à une fréquence moindre dans le cancer du côlon (35 à 43 % des tumeurs, codons 12 et 13), puis le cancer du poumon (20 à 30 %). La mutation est beaucoup plus rare au cours des cancers des voies biliaires (cholangiocarcinome) de l’endomètre, de l’ovaire, de l’estomac, de l’œsophage, et de la thyroïde [2]. L’adénocarcinome pancréatique se développe majoritairement à partir des cellules excréto-canalaires. Il est bien établi que son développement se fait à partir de lésions dites prénéoplasiques dont la présence constitue un facteur de risque de développement de ce cancer. En premier lieu, il s’agit des lésions néoplasiques pancréatiques intraépithéliales ou PanIN (pancreatic intraepithelial neoplasia). Il existe trois sous-types de PanIN. Les PanIN de type 1, subdivisées en type A et B, sont formées de cellules canalaires produisant une grande quantité de mucine intracytoplasmique et dont le noyau, petit, est maintenu au niveau de la membrane basale. Dans les PanIN de type 2, il existe une perte de la polarité du noyau et une élévation du rapport nucléocytoplasmique. Dans ce type, les lésions prennent un aspect papillaire complexe. Enfin, le type 3 se caractérise par de fortes atypies avec un noyau proéminent et des mitoses anormales ; il est considéré comme un carcinome in situ (Figure 2). Ces lésions sont reproduites au cours de la carcinogenèse expérimentale, comme le modèle chimio-induit chez le hamster doré de Syrie et les modèles transgéniques établis chez la souris.
 | Figure 2.
Fréquences des principales altérations géniques au sein des lésions précancéreuses pancréatiques humaines.
|
Les autres lésions prénéoplasiques sont constituées par les TIPMP (tumeurs intracanalaires papillaires mucineuses du pancréas) qui correspondent à une prolifération papillaire avec une muci-sécrétion de l’épithélium canalaire. Elles peuvent se développer aux dépens du canal pancréatique principal et/ou d’un canal secondaire (elles prennent dans ce cas un caractère kystique). Le risque de dégénérescence est variable selon que l’atteinte touche le canal de Wirsung (risque de 50 à 60 % à 5 ans) ou les canaux secondaires (risque de 5 à 15 % à 5 ans). Enfin, les cystadénomes mucineux sont aussi des lésions kystiques ne communiquant pas avec le canal pancréatique, constituées d’un épithélium dit « ovarien » sécrétant du mucus. Le risque de dégénérescence est évalué à 30 %. L’analyse génomique dans les lésions prénéoplasiques de type PanIN révèle une mutation Kras dans tous les cas : cette mutation apparaît précocement dès le stade PanIN de type 1, et la fréquence de la mutation augmente au fur à mesure de la progression des lésions (Figure 2) [
5,
6]. La mutation de Kras est aussi mise en évidence dans 50 % des TIPMP, tous stades anatomopathologiques confondus. Dans les cystadénomes mucineux, sa fréquence est variable selon l’histologie : en cas d’adénome, elle sera de 20 %, 33 % pour les lésions borderline et 87 % dans les carcinomes in situ. Outre la mutation activatrice de l’oncogène Kras, l’expression de gènes suppresseurs est altérée au cours de la carcinogenèse pancréatique : le gène CDKN2A qui code pour la protéine p16 (inhibiteur la cycline D/CDK4, protéine kinase), SMAD4 (mothers against decapentaplegic homolog 4)/DPC4, qui joue un rôle prépondérant dans la signalisation intracellulaire via la voie du TGFβ, et le gène TP53 (Figure 2). D’autres altérations géniques se produisent avec des mutations de l’EGFR et de BRCA2 [breast cancer 2], et l’activation de la télomérase [1–3]. Il ressort donc que la mutation de Kras est la plus fréquente et la plus précoce au cours du cancer pancréatique, ce qui a suscité de nombreux travaux au plan de la recherche fondamentale, mais aussi de la recherche clinique. |
Applications en recherche Les possibilités d’expression hétérologue du gène Kras dans des modèles cellulaires ont permis d’entreprendre des travaux de carcinogenèse in vitro, et l’équipe de Weinberg a ouvert la voie en démontrant que la carcinogenèse humaine, et notamment l’émergence des altérations génétiques, était de type « multi-étapes » [
7]. Cette équipe avait confirmé que la transformation maligne de cellules humaines ne se produisait qu’après activation mono-allélique d’oncogènes et mutation/délétion bi-allélique de gènes suppresseurs de tumeurs, et ce avec une cinétique bien précise. L’activation de Kras par mutation faisait partie de cette cascade. Dans un autre registre, la connaissance de la mutation de l’oncogène Kras et ses conséquences en termes de prolifération et d’activation des systèmes de transduction sont importantes pour décortiquer les mécanismes de prolifération et de progression tumorale de la cellule. En effet, l’inactivation de la voie Ras sera nécessaire pour faire la part des événements dépendants de Ras et de ceux relevant des autres voies de régulation intracellulaires. Enfin, les cellules présentant des mutations de Kras seront autant de modèles in vitro pour tester des molécules à visées thérapeutiques, comme les inhibiteurs des farnésyltransférases, les inhibiteurs antisens, et les siARN (petits ARN interférents). À côté des modèles cellulaires, les modèles transgéniques animaux ont ouvert une nouvelle voie dans l’étude de la carcinogenèse pancréatique expérimentale. Les premiers modèles murins ont ciblé l’expression constitutive de l’oncogène Kras dans les acinus pancréatiques grâce au promoteur de l’élastase ou par insertion dans le gène de Mist1 (muscle, intestine and stomach expression 1), un facteur contrôlant le phénotype acinaire. Kras a aussi été exprimé dans des cellules progénitrices pancréatiques par knock-in dans le gène de la nestine. Ces modèles ont produit des lésions cancéreuses majoritairement acinaires et/ou des lésions prénéoplasiques qui n’ont pas progressé jusqu’au stade cancer. De façon inattendue, le ciblage de Kras avec le promoteur de la cytokératine 19, un marqueur des cellules canalaires, n’a produit ni tumeur, ni lésions précancéreuses. En 2003, les souris KC ont permis de franchir une étape importante. La mutation KrasG12D
est introduite après une cassette Lox-Stop-Lox (LSL), empêchant la transcription, dans un allèle du gène de Ras. La version oncogénique de Kras est alors induite, à des niveaux physiologiques, pendant le développement embryonnaire après excision de la cassette LSL par la recombinase Cre exprimée sous le contrôle des promoteurs des facteurs de transcription Pdx1 (pancreatic and duodenal homeobox 1) ou Ptf1 (pancreatic transcription factor 1), spécifiques des cellules souches pancréatiques [
8,
9] (Tableau I).
Tableau I.
|
Lésions précancéreuses |
Tumeurs |
| Génotypes |
PanIN |
TIPMP |
CM |
Âge (mois) |
ADK |
Métastases |
Références |
| Pdx1-Cre ; Kras
G12D
|
Oui |
|
|
> 12 |
D |
Oui |
[8] |
| Ptf1a-Cre ; Kras
G12D
|
Oui |
|
|
> 12 |
D |
Oui |
[8] |
| Ptf1a-Cre ; Kras
G12D ; Ela-TGFα |
Oui |
Oui |
|
5 |
D |
Oui |
[9] |
| Pdx1-Cre ; Kras
G12D ; Ink4a/Arflox/lox
|
Oui |
|
|
2 |
I |
Non |
[
10] |
| Pdx1-Cre ; Kras
G12D ; p53lox/lox
|
Oui |
|
|
1,5 |
D |
Non |
[
11] |
| Pdx1-Cre ; Kras
G12D ; p53R127H/+
|
Oui |
|
|
2,5 |
D |
Oui |
[
12] |
| Pdx1-Cre ; Kras
G12D ; Smad4lox/lox
|
|
Oui |
|
4 |
D |
Oui |
[
13] |
| Ptf1a-Cre ; Kras
G12D ; Smad4lox/lox
|
|
|
Oui |
3,5 |
D |
Oui |
[
14] |
| Ptf1a-Cre ; Kras
G12D ; p53R127H/+ ; Brca2Tr/D11
|
Oui |
|
|
2 |
D |
Oui |
[
15] |
| Ela-tTA TRE-Cre ; Kras
G12V ± pancréatite |
Oui |
|
|
8-12 |
D |
Non |
[
16] |
Modèles animaux transgéniques développés à partir de la mutation de l’oncogène Kras. PanIN : pancreatic intraepithelial neoplasia ; TIPMP : tumeurs intrapapillaires et mucineuses du pancréas ; CM : cystadénome mucineux ; ADK : adénocarcinome pancréatique ; D : différencié ; I : indifférencié ; Pdx1 : pancreatic and duodenal homeobox 1 ; Ptf1a : pancreatic transcription factor 1a ; Ela : élastase ; TGFα : transforming growth factor a ; tTA : tetracyclin transactivator ; TRE : tetracyclin response element. |
En moins d’une décennie, ces souris génétiquement modifiées ont pris une place prépondérante comme modèles de cancer du pancréas. Elles miment fidèlement la pathologie humaine : 100 % des souris reproduisent les différentes lésions précancéreuses de type PanIN, du stade PanIN1A au stade PanIN3 (Figure 3). Les PanIN murines sont similaires aux PanIN humaines du point de vue des voies de signalisation activées et des protéines exprimées. Ces souris confirment le rôle initiateur essentiel joué par l’oncogène Kras dans la carcinogenèse pancréatique. Une faible proportion (10 %) des souris développent des tumeurs de phénotype canalaire à un âge avancé, ce qui suggère la nécessité d’un événement additionnel pour que les lésions précancéreuses progressent jusqu’au stade cancer. Sur la base des caractéristiques moléculaires des tumeurs pancréatiques humaines, un panel important de modèles a été généré en combinant la mutation de Kras avec d’autres altérations génétiques. L’inactivation de gènes suppresseurs de tumeurs de souris, comme TP53, SMAD4, et INK4A/ARF, mutés dans la pathologie humaine, conduit au développement, en quelques mois, de tumeurs invasives entourées d’un stroma dense. Il est intéressant de noter que le phénotype des tumeurs et des lésions prénéoplasiques varie selon le génotype. Par exemple, la délétion de Smad4 dans le pancréas d’une souris KC induit le développement de TIPMP et de cystadénomes mucineux, alors que l’expression du TGFα dans ces souris génère des TIPMP (Tableau I) [10–14]. Pour reproduire au mieux la pathologie humaine, la mutation de Kras a été induite à l’âge adulte. Certains mécanismes d’initiation du cancer, ainsi que le lien entre inflammation et oncogenèse pancréatique ont été démontrés et analysés en soumettant ces souris à des protocoles induisant une pancréatite chronique [15, 16].
 | Figure 3.
Coupe histologique montrant la présence de lésions pré-cancéreuses de type PanIN dans le pancréas des souris transgéniques Pdx1-Cre ; LSL-KrasG12D. A : cellules acinaires ; I : îlot de Langerhans ; C : canal. Les foyers de lésions de dysplasie de bas grade sont entourés ; une lésion de dysplasie de plus haut grade formée à partir d’un canal est signalée par un astérisque. |
Les applications et perspectives qu’offrent les modèles murins dérivés des souris KC dans les domaines de la prévention, du diagnostic [
17] et de la thérapie sont larges. De même, des méthodes d’imagerie, comme le PET-CT (imagerie par émission de positons), ont été testées pour le dépistage des lésions PanIN dans ce modèle [
18]. Enfin, ce modèle a aussi permis de démontrer l’importance de la barrière physique que constitue le microenvironnement tumoral qui s’oppose aux stratégies d’augmentation de l’efficacité de la chimiothérapie [
19]. |
Applications en clinique humaine Rôle dans le diagnostic Des techniques de détection ont été développées pour l’analyse des mutations de Kras dans les différents milieux biologiques humains. Elles prennent en compte le fait que la mutation de l’oncogène Kras est fréquente au cours du cancer du pancréas, est bien caractérisée et présente sous forme de hotspots, notamment sur le codon 12. Elles sont basées sur les progrès d’extraction d’ADN (avec utilisation de microkits) et de l’amplification génique, qui ont grandement facilité ce développement clinique. Techniques de détection La plus ancienne est l’analyse par RFLP ( restriction fragment length polymorphism) qui est basée sur le caractère unique du site de la mutation en permettant l’utilisation d’une enzyme de restriction (BstNl pour la mutation du codon 12) [ 4,
20]. L’autre méthode couramment appliquée est le séquençage direct après PCR ( polymerase chain reaction) au moyen d’amorces encadrant la région des codons 12 et 13 [ 20]. D’autres méthodes sont actuellement moins utilisées : l’hybridation DDGE ( denaturating gel gradient electrophoresis) ou SSCP ( single strand conformation polymorphism). Plus récemment ont émergé la technique du snapshot (SNP) et de la PCR quantitative en temps réel. Le séquençage direct, le SNP et la PCR en temps réel avec discrimination allélique sont actuellement les méthodes les plus compétitives en termes de coût et de fiabilité. Études sur les milieux biologiques Leur objectif est d’essayer d’améliorer le diagnostic du cancer du pancréas. Il s’agit d’affirmer le diagnostic devant des symptômes (douleurs abdominales et amaigrissement, pancréatite aiguë apparemment idiopathique) ou un contexte évocateurs, alors que l’imagerie est non contributive. Il s’agit aussi du diagnostic différentiel dans le cas d’une pancréatite chronique à forme pseudotumorale, c’est-à-dire chez des patients atteints d’une pancréatite chronique avec une masse tissulaire pancréatique. Ce problème crucial en pancréatologie clinique a été abordé en recherchant la mutation Kras dans divers milieux biologiques, comme le sang circulant, le suc pancréatique pur, le liquide duodénal, et le matériel de ponction pancréatique. Beaucoup d’études ont initialement (avant les années 2000) concerné le suc pur prélevé par cholangiopancréatographie par voie rétrograde per-endoscopique (CPRE) chez des patients porteurs de cancer et de pancréatite chronique. Cette technique endoscopique n’est actuellement plus appliquée en clinique dans un but diagnostique, mais uniquement dans un but thérapeutique. En effet, cet examen invasif a été remplacé par l’imagerie en coupe, comme le scanner ou l’IRM (imagerie par résonance magnétique), mais aussi par l’écho-endoscopie. Si l’on cumule les études de CPRE, près de 500 patients porteurs de cancer et 300 de pancréatite chronique ont été évalués. La sensibilité de la recherche de la mutation de Kras (le plus souvent par RFLP suivi d’un séquençage) était de 73 %, et la spécificité était de 90 %. Malheureusement cette recherche n’a pas permis le diagnostic de formes débutantes de cancer du pancréas [4,
21]. D’autre part, l’analyse concomitante d’autres altérations géniques dans le suc (P16, DPC4, TP53) n’a pas amélioré ces performances [
22]. En revanche, le suivi à long terme des patients porteurs de pancréatite chronique et d’une mutation de l’oncogène Kras (10 % des patients étudiés) détectée dans le suc n’a pas révélé d’apparition ultérieure de cancer du pancréas, confirmant peut être le caractère « multi-étapes » de la carcinogenèse pancréatique. En d’autres termes, d’autres altérations géniques sont nécessaires pour une carcinogenèse avérée avec probablement une progression PanIN-adénocarcinome. Les études effectuées sur le liquide duodénal, le plasma ou les selles sont moins nombreuses : leur sensibilité est plus faible (entre 40 et 60 %) et leur spécificité toujours élevée, de 90 à 95 %, en ce qui concerne la recherche de la mutation de Kras pour le diagnostic de cancer du pancréas. Études sur le matériel de ponction pancréatique Depuis l’avènement de l’écho-endoscopie et des cytoponctions dirigées, le diagnostic des lésions tissulaires pancréatiques a été grandement amélioré. Néanmoins, la sensibilité de l’examen cytopathologique pour le diagnostic de cancer oscille entre 65 et 95 %, avec une valeur prédictive négative allant de 50 à 70 %. La spécificité est assez élevée (entre 90 et 100 %), ce qui permet d’assurer le diagnostic de bénignité des masses pancréatiques. Plusieurs études, incluant celle menée par notre équipe, ont évalué l’importance de la recherche de la mutation de Kras dans le matériel de cytoponction pancréatique pour le diagnostic d’adénocarcinome (Tableau II) [ 20,
23–
26]. Cette analyse apporte, tout d’abord, une contribution au diagnostic différentiel entre cancer et pancréatite chronique pseudotumorale. En effet, devant un tableau de pancréatite chronique avec masse tissulaire pancréatique, l’absence de mutation de Kras dans les cellules obtenues par ponction est fortement en faveur de la bénignité. En revanche, en cas de ponction non contributive par absence de cellules malignes évidentes et d’un gène Kras muté, le diagnostic de malignité d’une masse tissulaire pancréatique est hautement probable (impliquant une nouvelle biopsie et/ou une chirurgie). La recherche de la mutation de Kras dans le matériel de cytoponction obtenu sous écho-endoscopie serait donc un complément au diagnostic cytopathologique. Même si la faible cellularité du matériel de cytoponction peut altérer la sensibilité de l’analyse de Kras, cette dernière apparaît tout de même utile et complémentaire de la cytologie. L’analyse de Kras dans le liquide de ponction semble aussi très utile dans le cadre du diagnostic de malignité des cystadénomes mucineux et des TIPMP, mais nous disposons de moins d’études dans ce domaine.
Tableau II.
| Nombrede patientscancer/PCC |
Sensibilité (%) |
Spécificité (%) |
Précision (%) |
Réf. |
| 28/8 |
Kras : 77CytoP : 62CytoP + Kras : 81 |
Kras : 100CytoP : 100CytoP + Kras : 100 |
Kras : 82CytoP : 71CytoP + Kras : 85 |
[23] |
|
| 33/24 |
Kras : 73CytoP : 97CytoP + Kras : 97 |
Kras : 100CytoP : 100CytoP + Kras : 100 |
Kras : 91CytoP : 84CytoP + Kras : 98 |
[
24] |
|
| 62/15 |
Kras : 74CytoP : 84CytoP + Kras : 94 |
Kras : 100CytoP : 100CytoP + Kras : 100 |
-CytoP : 58- |
[
25] |
|
| 57/11 |
CytoP : 82CytoP + Kras : 90 |
CytoP : 97CytoP + Kras : 47 |
CytoP : 59CytoP + Kras : 89 |
[26] |
|
| 129/27 |
Kras : 67CytoP : 83CytoP + Kras : 88 |
Kras : 100CytoP : 100CytoP + Kras : 100 |
Kras : 86CytoP : 72CytoP + Kras : 90 |
[20] |
Évaluation des performances de la recherche de la mutation de l’oncogène Kras dans le matériel de cytoponction prélevé sous écho-endoscopie chez des patients atteints de cancers du pancréas et de pancréatites chroniques. PCC : pancréatite chronique calcifiante ; CytoP : cytopathologie. |
Rôle possible dans le traitement et le pronostic du cancer du pancréas Le développement d’inhibiteurs des farnésyltransférases a débouché sur des essais incluant des patients porteurs d’adénocarcinomes pancréatiques localement avancés. Il n’a pas été mis en évidence d’effet notable. Il en est de même pour les stratégies de vaccination à base de mélanges de peptides p21-Ras porteurs des différentes mutations. Des études de phase 1 sont en cours pour tester la stratégie ARN interférents dans cette indication. En ce qui concerne l’évaluation de Kras comme facteur pronostique dans le cancer du pancréas, plusieurs études ont été menées chez des patients opérés, mais aussi chez des patients porteurs de tumeurs non résécables et/ou métastatiques. Les résultats de ces différentes études sont d’interprétation difficile compte tenu de l’hétérogénéité des populations et du faible nombre de cas [
27]. En ce qui concerne les adénocarcinomes réséqués, la présence d’une mutation de Kras est très certainement un facteur de mauvais pronostic. En revanche, ceci reste à prouver pour les cancers localement avancés [ 27,
28]. |
Depuis la mise en évidence, il y a plus de 30 ans, de l’oncogène Kras et de son statut de mutation prédominante dans le cancer du pancréas, la recherche biomédicale a profité de cette découverte à plusieurs titres : la compréhension des phénomènes de prolifération et d’invasion non contrôlées de la cellule cancéreuse pancréatique, le développement de modèles transgéniques reproduisant la carcinogenèse pancréatique humaine, et le développement d’outils pour la recherche de la mutation de Kras applicables en pratique clinique quotidienne. Si ces outils sont très utiles dans le cadre de la prise en charge des cancers colorectaux métastatiques [
32] (contre-indication d’un traitement par anti-EGFR en présence de la mutation de Kras) [
29,
30], on entrevoit quelques applications dans le cadre du cancer du pancréas : aide au diagnostic des masses solides pancréatiques, voire appréciation du pronostic. Les implications dans le traitement ne sont pas encore bien appréhendées. Il n’en reste pas moins que les études portant sur Kras à l’échelle cellulaire, dans des modèles animaux et dans les tumeurs et tissus humains, ont apporté un lot inestimable de connaissances avec, in fine, des applications cliniques quotidiennes. C’est un excellent exemple de transfert de technologie de la recherche fondamentale vers la clinique humaine et les soins médicaux. |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
|
Footnotes |
1.
Hidalgo
M
. Pancreatic cancer . N Engl J Med.
2010; ; 362 : :1605.–1617. 3.
Torrisani
J
,
Buscail
L
. Molecular pathways of pancreatic carcinogenesis . Ann Pathol.
2002; ; 22 : :349.–355. 4.
Berthélemy
P
,
Bouisson
M
,
Escourrou
J
, et al.
Identification of KRAS mutations in pancreatic juice in the early diagnosis of pancreatic cancer . Ann Intern Med.
1995; ; 123 : :188.–191. 5.
Laghi
L
,
Orbetegli
O
,
Bianchi
P
, et al.
Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers . Oncogene.
2002; ; 21 : :4301.–4306. 6.
Hruban
RH
,
Wilentz
RE
,
Kern
SE
. Genetic progression in the pancreatic ducts . Am J Pathol.
2000; ; 156 : :1821.–1825. 7.
Hahn
WC
,
Counter
CM
,
Lundberg
AS
, et al.
Creation of human tumour cells with defined genetic elements . Nature.
1999; ; 400 : :464.–468. 8.
Hingorani
SR
,
Petricoin
EF
,
Maitra
A
, et al.
Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse . Cancer Cell.
2003; ; 4 : :437.–450. 9.
Siveke
JT
,
Einwachter
H
,
Sipos
B
, et al.
Concomitant pancreatic activation of Kras(G12D) and Tgfα results in cystic papillary neoplasms reminiscent of human IPMN . Cancer Cell.
2007; ; 12 : :266.–279. 10.
Aguirre
AJ
,
Bardeesy
N
,
Sinha
M
, et al.
Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma . Genes Dev.
2003; ; 17 : :3112.–3126. 11.
Bardeesy
N
,
Aguirre
AJ
,
Chu
GC
, et al.
Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse . Proc Natl Acad Sci USA.
2006; ; 103 : :5947.–5952. 12.
Hingorani
SR
,
Wang
L
,
Multani
AS
, et al.
Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice . Cancer Cell.
2005; ; 7 : :469.–483. 13.
Kojima
K
,
Vickers
SM
,
Adsay
NV
, et al.
Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia . Cancer Res.
2007; ; 67 : :8121.–8130. 14.
Izeradjene
K
,
Combs
C
,
Best
M
, et al.
Kras(G12D) and Smad4/DPC4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas . Cancer Cell.
2007; ; 11 : :229.–243. 15.
Skoulidis
F
,
Cassidy
LD
,
Pisupati
V
, et al.
Germline BRCA2 heterozygosity promotes Kras(G12D)-driven carcinogenesis in a murine model of familial pancreatic cancer . Cancer Cell.
2010; ; 18 : :499.–509. 16.
Guerra
C
,
Schuhmacher
AJ
,
Canamero
M
, et al.
Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice . Cancer Cell.
2007; ; 11 : :291.–302. 17.
Faca
VM
,
Song
KS
,
Wang
H
, et al.
A mouse to human search for plasma proteome changes associated with pancreatic tumor development . PLoS Med.
2008; ; 5 : :e123.. 18.
Fendrich
V
,
Schneider
R
,
Maitra
A
, et al.
Detection of precursor lesions of pancreatic adenocarcinoma in PET-CT in a genetically engineered mouse model of pancreatic cancer . Neoplasia.
2011; ; 13 : :180.–186. 19.
Olive
KP
,
Jacobetz
MA
,
Davidson
CJ
, et al.
Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer . Science.
2009; ; 324 : :1457.–1461. 20.
Bournet
B
,
Souque
A
,
Senesse
P
, et al.
EUS-guided fine needle-aspiration biopsy coupled to KRAS mutation assay to differentiate pancreatic cancer from pseudo-tumorous chronic pancreatitis . Endoscopy.
2009; ; 41 : :552.–557. 21.
Van Laethem
JL
,
Vertongen
P
,
Deviere
J
, et al.
Detection of c-Ki-ras gene codon 12 mutations from pancreatic duct brushings in the diagnosis of pancreatic tumours . Gut.
1995; ; 36 : :781.–787. 22.
Costentin
L
,
Pagès
P
,
Bouisson
M
, et al.
Frequent deletions of tumor suppressor genes in pure pancreatic juice from patients with tumoral or nontumoral pancreatic diseases . Pancreatology.
2002; ; 2 : :17.–25. 23.
Tada
M
,
Komatsu
Y
,
Kawabe
T
, et al.
Quantitative analysis of K-ras gene mutation in pancreatic tissue obtained by endoscopic ultrasonography-guided fine needle aspiration : clinical utility for diagnosis of pancreatic tumor . Am J Gastroenterol.
2002; ; 97 : :2263.–2270. 24.
Pellisé
M
,
Castells
A
,
Ginès
A
, et al.
Clinical usefulness of KRAS mutational analysis in the diagnosis of pancreatic adenocarcinoma by means of endosonography-guided fine-needle aspiration biopsy . Aliment Pharmacol Ther.
2003; ; 17 : :1299.–1307. 25.
Takahashi
K
,
Yamao
K
,
Okubo
K
, et al.
Differential diagnosis of pancreatic cancer and focal pancreatitis by using EUS-guided FNA . Gastrointest Endosc.
2005; ; 61 : :76.–79. 26.
Maluf-Filho
F
,
Kumar
A
,
Gerhardt
R
, et al.
Kras mutation analysis of fine needle aspirate under EUS guidance facilitates risk stratification of patients with pancreatic mass . J Clin Gastroenterol.
2007; ; 41 : :906.–910. 27.
Bournet
B
,
Muscari
F
,
Guimbaud
R
, et al.
KRAS mutations and their correlation with survival of patients with advanced pancreatic cancer . Pancreas.
2013; ; 42 : :543.–544. 28.
Shin
SH
,
Kim
SC
,
Hong
S-M
, et al.
Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival . Pancreas.
2013; ; 42 : :216.–222. 29.
Lièvre
A
,
Bachet
JB
,
Le Corre
D
, et al.
KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer . Cancer Res.
2006; ; 66 : :3992.–3995. 30.
Laurent-Puig
P
. Critères biologiques d’éligibilité pour un traitement anti-EGFR . Med Sci (Paris).
2009; ; 25 : :21.–24. 31.
Galmiche
A
,
Ezzoukhry
Z
. Régulation de la survie cellulaire par les kinases de la famille RAF . Med Sci (Paris).
2010; ; 26 : :729.–733. 32.
Viret
F
,
Gonçalves
A
. Cancers colorectaux métastatiques et thérapies ciblées anti-EGF . Med Sci (Paris).
2009; ; 25 : :13.–21. 33.
Ahearn
IM
,
Haigis
K
,
Bar-Sagi
D
,
Philips
MR
. Regulating the regulator: post-translational modification of Ras . Nat Rev Mol Cell Biol.
2012; ; 13 : :39.–51. |




