| |
| Med Sci (Paris). 2014 October; 30(10): 874–881. Published online 2014 October 14. doi: 10.1051/medsci/20143010014.Les lymphocytes B Une cible prometteuse pour traiter l’athérosclérose ? Moustafa Hamze,1,2* Caroline Desmetz,1** and Paul Guglielmi3*** 1Centre de pharmacologie et d’innovation dans le diabète, EA 7288, Faculté de pharmacie, 15, avenue Charles Flahault, 34093Montpellier, France 2Institut de biologie et de technologies de Saclay (iBiTec-S), service d’ingénierie moléculaire des protéines (SIMOPRO), centre CEA Saclay-Gif-sur-Yvette, 91191Gif-sur-Yvette Cedex, France 3Dynamique des interactions membranaires normales et pathologiques (DIMNP), UMR 5235 CNRS, Université Montpellier II, place Eugène Bataillon, 34095Montpellier Cedex 5, France |
Les maladies cardiovasculaires sont un véritable problème de santé publique. Elles sont la première cause de mortalité dans le monde – 13,2 millions de décès sont imputables aux cardiopathies ischémiques et aux accidents cardiovasculaires cérébraux en 2011 selon l’Organisation mondiale de la santé (OMS) [
1]. Bien que la mortalité attribuable aux maladies cardiovasculaires diminue, leur incidence augmente significativement [
2]. L’athérosclérose est la principale cause des maladies cardiovasculaires. Elle se traduit par des pathologies chroniques (angor d’effort) et des maladies aiguës souvent mortelles (infarctus du myocarde et accident vasculaire cérébral). L’athérosclérose est actuellement définie comme une maladie inflammatoire multifactorielle et progressive qui commence dès la vie fœtale [1]. Elle implique des facteurs génétiques autant qu’hémodynamiques, métaboliques, comportementaux et environnementaux [
3]. L’accumulation de lipoprotéines oxydées (LDLox, oxidized low-density lipoprotein) dans l’espace sous-endothélial artériel (intima du vaisseau) constitue l’étape initiale de formation des plaques d’athérosclérose. Les LDLox activent les cellules endothéliales, induisant l’adhérence et le recrutement des cellules immunitaires (Figure 1). Les macrophages pariétaux captent les LDLox et se transforment en cellules spumeuses qui s’accumulent pour former des stries lipidiques [1,
4]. Ils sécrètent alors des cytokines, des facteurs de croissance et des enzymes qui augmentent l’instabilité des plaques.
 | Figure 1.
Cellules immunitaires dans une artère normale ou pathologique. Le délai de constitution des lésions d’athérome est long (des décénnies chez l’homme) et la structure de ces lésions varie avec le temps. Les lésions précoces (stries lipidiques) se forment dans la couche interne du vaisseau (intima) qui contient un grand nombre de macrophages chargés en lipides (cellules spumeuses). Les lésions évoluées (plaques athéromateuses matures) ont une structure plus complexe : autour d’une région centrale contenant des cellules spumeuses et des gouttelettes lipidiques se trouvent des cellules musculaires lisses et un réseau de fibres de collagène. Une chape fibreuse sous-endothéliale contribue à la stabilité de la plaque et isole le noyau lipidique de la lumière artérielle. Les cellules du système immunitaire présentes comportent majoritairement des lymphocytes T et des macrophages, mais aussi des cellules dendritiques, des mastocytes et des cellules T natural killer (NK). Les lymphocytes B sont présents en faible nombre au sein des plaques d’athérome, mais sont retrouvés à distance dans la tunique extérieure de l’artère (adventice) et les ganglions lymphatiques drainants. Ces lésions peuvent devenir instables par fracture de la chape fibreuse, mettant en contact les protéines circulantes de la coagulation avec le facteur tissulaire du noyau lipidique et aboutissant à des thromboses occlusives aiguës responsables des complications cliniques les plus graves. Une progression régulière des lésions représente l’exception plutôt que la règle. |
Le système immunitaire adaptatif intervient également dans le développement de l’athérome [
31]. Les lymphocytes T (LT) sont présents dans les plaques à tous les stades de la maladie. Les lymphocytes T activés (TCR [T cell receptor] αβ+ CD4+) constituent la population lymphocytaire dominante des parois artérielles malades et sont pour la plupart de type auxiliaire 1 (Th1). Les lymphocytes T régulateurs (Treg), naturels ou induits, inhibent efficacement le développement de l’athérosclérose expérimentale dans plusieurs modèles animaux, alors que l’appauvrissement en Treg aggrave la formation de plaques. Le rôle des lymphocytes T dans le contrôle du processus athéromateux a initialement été considéré comme dominant. Cependant, la participation des lymphocytes B à cette pathologie a été récemment réexaminée [
5]. Ces derniers peuvent, outre leur production d’anticorps, moduler l’activité des lymphocytes T, présenter des antigènes, sécréter des cytokines et participer à la formation de structures lymphoïdes tertiaires dans les parois vasculaires, contribuant ainsi à l’évolution des plaques [
6]. La démarche thérapeutique dans l’athérosclérose est fondée sur la réduction de la charge lipidique. Certains des médicaments utilisés à cette fin ont aussi des effets anti-inflammatoires sur la paroi artérielle. Ces traitements ralentissent la progression de la maladie (30-40 %), mais sont incapables d’inhiber complètement la formation des plaques [
7]. Une meilleure connaissance des mécanismes physiopathologiques et inflammatoires permettrait de lutter plus efficacement contre la constitution des lésions vasculaires et leurs complications. |
Implication des lymphocytes B dans l’athérosclérose La participation de l’immunité humorale au processus athérosclérotique a d’abord été mise en évidence par l’augmentation des taux d’anticorps et de complexes immuns dans le sérum des patients atteints [1]. L’intérêt pour les autres propriétés des lymphocytes B ne s’est développé que récemment. Les études des fonctions des lymphocytes B dans des modèles expérimentaux ont parfois abouti à des résultats contradictoires (Tableau I). Caligiuri et al. ont été les premiers à préciser le rôle de ces cellules dans l’athérosclérose : la splénectomie aggrave la maladie des souris déficientes en apolipoprotéine E (Apoe
-/-
, un modèle robuste d’athérogenèse), alors que le transfert de lymphocytes B spléniques purifiés est athéro-protecteur, réduisant la taille des lésions post-splénectomie [
8]. Ce résultat a été confirmé par l’aggravation des lésions vasculaires dans le modèle des souris déficientes à la fois en récepteur des LDL et en lymphocytes B (souris Ldlr
-/-
μMT) [
9]. Un des mécanismes de l’effet protecteur de ces lymphocytes est lié à la production d’anticorps dirigés contre les LDLox, qui accélèrent la clairance de ces lipoprotéines délétères. De nombreux anticorps anti-LDLox sont des anticorps naturels (Encadré 1).
Tableau I.
| Modèles |
Effets attribués aux LB |
Sous-populations de LB concernées |
Références |
| Splénectomie des souris Apoe
-/-
|
|
|
[8] |
| Transfert de splénocytes |
Protecteur |
Non identifiées |
[8] |
| Souris Ldlr
-/-
déficientes en cellules B |
|
|
[9] |
|
| Déplétion des cellules B chez les souris Apoe
-/-
et Ldlr
-/-
|
Pathogène |
Non identifiées |
[
10,
11] |
|
| Splénectomie des souris Apoe
-/-
|
|
|
[
14] |
| Transfert des cellules B1 |
Protecteur |
B1 |
[14] |
| Souris Ldlr
-/-
IgM
-/-
|
|
|
[
21] |
|
| Déplétion des cellules B chez les souris Apoe
-/-
|
|
|
[10] |
| Transfert des cellules B2 à des souris Apoe
-/-
RAG
-/-
|
Pathogène |
B2 |
[11] |
| Anti-BAFFR et souris déficientes en BAFFR (BAFFR-/-) |
|
|
[14,
20] |
Rôle des lymphocytes B dans les modèles expérimentaux d’athérosclérose. RAG : recombination-activating gene; BAFFR : récepteur BAFF; LB : lymphocytes B. |
|
1. Anticorps naturels Les anticorps naturels sont produits par les lymphocytes B1 en l’absence de stimulation par les antigènes et de coopération avec les lymphocytes T. Ils appartiennent le plus souvent à la classe IgM. Ils utilisent des séquences de régions variables en configuration germinale et n’ont pas recours à l’hypermutation somatique. Le répertoire anticorps résultant est restreint et stéréotypé mais suffisamment large pour reconnaître de nombreuses spécificités. Parmi celles-ci se trouvent des antigènes du soi, du soi modifié et des antigènes étrangers, comme certains phospholipides, des déterminants glucidiques, l’ADN simple brin, des peptides ou des glycoprotéines membranaires. Ils constituent donc une première ligne de défense contre les agents pathogènes et participent aussi à l’homéostasie tissulaire en favorisant l’élimination des molécules altérées et des cellules apoptotiques ou nécrotiques. Une proportion appréciable des IgA (surtout au cours de la période néonatale) fait aussi partie des anticorps naturels et exerce ainsi une fonction anti-inflammatoire.
|
Cependant, des travaux récents dans un modèle animal ont montré que l’administration d’anticorps anti-CD20, éliminant les lymphocytes B en ciblant une de leur molécule membranaire (Encadré 2), s’accompagnait d’une réduction de l’atteinte vasculaire [10, 11]. Ce paradoxe s’explique par le fait que la splénectomie d’une part, et l’anticorps anti-CD20 d’autre part, ciblent des sous-populations différentes de lymphocytes B qui exercent des effets opposés durant la maladie athéromateuse (respectivement les lymphocytes B1, proches de l’immunité innée, et les lymphocytes conventionnels B2,
Encadré 3
).
|
2. Antigène CD20 L’antigène de surface CD20 est une phosphoprotéine glycosylée qui est exprimée à tous les stades de différenciation des lymphocytes B, à l’exception des précurseurs B (lymphocytes pro-B précoces) et des cellules les plus matures (plasmablastes et plasmocytes). L’antigène CD20 est spécifique de la lignée lymphocytaire B, participe à la régulation des transports transmembranaires de calcium et intervient dans la progression du cycle cellulaire après activation des lymphocytes B. La molécule CD20 est la cible d’anticorps monoclonaux utilisés pour le traitement des néoplasies lymphocytaires B et de maladies inflammatoires auto-immunes (polyarthrite rhumatoïde, etc.) [
34]. Ces anticorps agissent par cytotoxicité dépendante du système du complément ou du recrutement de cellules tueuses. Une faible densité membranaire d’antigène CD20, une sélection de molécules CD20 mutantes ou une localisation des cellules cibles dans des niches protectrices (lymphocytes B1 péritonéaux) permettent à certains lymphocytes B d’échapper à la toxicité des anticorps anti-CD20.
|
|
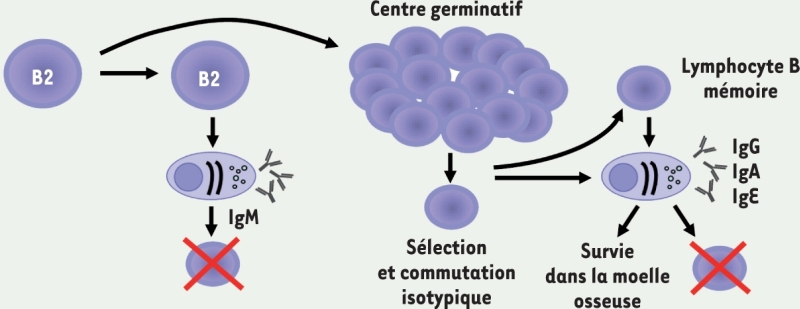
3.. Lymphocytes B Lymphocytes B1
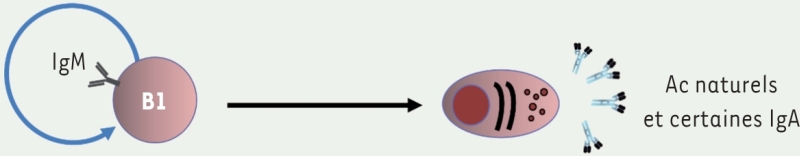
Leurs caractéristiques fonctionnelles les rendent proches de l’immunité innée. Développement durant la vie fœtale et post-natale, puis maintien par autorenouvellement. Localisation : cavités péritonéale et pleurale, lamina propria de l’intestin. Production d’Ig naturelles rapidement disponibles. Diversité limitée du répertoire anticorps. Ne deviennent généralement pas des lymphocytes « mémoire ». Fonction : protection précoce, rapide contre les infections virales et bactériennes. Marqueurs membranaires : CD27
-
CD5
+
IgM
+
IgD
+
ou CD20
+
CD27
+
CD43
+
CD70
-
(homme); B220
low
IgM
high
CD43
+
CD11b
+
CD5
+
(souris). Lymphocytes B2
Prototypes de l’immunité adaptative, souvent au repos avant la rencontre avec l’antigène. Localisation : peuplent les organes lymphoïdes secondaires, les muqueuses. Fonction : production d’Ig dépendante d’une interaction avec les lymphocytes T dans les centres germinatifs permettant l’hypermutation somatique et les commutations de classe des anticorps (IgG, IgA et IgE). Grande diversité du répertoire et maturation d’affinité des Ig. Possibilité de devenir des lymphocytes « mémoire ». Marqueurs membranaires : CD27
-
CD5
-
CD20
+
IgM
+
IgD
+
(homme); B220
+
CD5
-
CD23
+
CD21
+
(souris). Lymphocytes B régulateurs
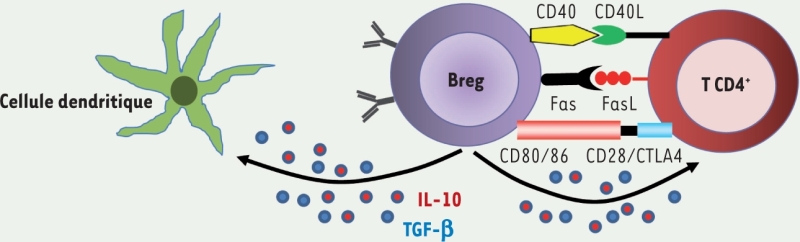
Effet atténuateur de l’inflammation dans de nombreuses pathologies auto-immunes expérimentales : encéphalite auto-immune, maladies inflammatoires chroniques du tube digestif, diabète de type 1 des souris NOD (non-obese diabetic), arthrite au collagène, hypersensibilité de contact. Les lymphocytes B régulateurs ont aussi un rôle dans l’immunité antitumorale et antiparasitaire : tolérance de tumeurs, infection par leishmania. Producteurs de cytokines IL-10 (maladies auto-immunes) ou de TGF-β (néoplasies). Peuvent, selon les contextes expérimentaux, provenir de lymphocytes B1 ou B2. Marqueurs membranaires : nombreux phénotypes contradictoires décrits dans les pathologies animales auto-immunes. Pas de consensus chez l’homme, bien que des propriétés régulatrices aient été documentées dans plusieurs sous-populations de lymphocytes B. CTLA4 : cytotoxic T-lymphocyte A antigen 4.
|
Des populations lymphocytaires B régulatrices (Breg), définies par leur capacité de production de la cytokine anti-inflammatoire interleukine (IL)-10 (Encadré 3), ont été décrites dans des modèles animaux de maladies auto-immunes (→) [6,
32]. L’absence de marqueur phénotypique spécifique n’a cependant pas encore permis de démontrer la participation des Breg à la physiopathologie de l’athérosclérose.
(→) Voir la Nouvelle de J.D. Bouaziz et al., m/s 2014, n° 8–9, page 721
|
L’immunité locale dans l’athérosclérose La présence de dépôts d’IgM (immunoglobulines M) et d’IgG anti-LDLox au sein des lésions humaines [
13] a incité plusieurs groupes à caractériser la réponse immune locale [1]. De nombreux types de cellules immunitaires innées et des lymphocytes T sont détectés dans les parois artérielles humaines et murines dès les phases précoces de l’athérosclérose (Figure 1). Les lymphocytes B sont présents dans les lésions primaires de la souris, mais plutôt lors des phases avancées de l’athérosclérose humaine [
12]. L’importance de l’infiltration de la tunique extérieure de l’artère (adventice) par les lymphocytes B est corrélée à la sévérité de la maladie [14]. Les cellules immunitaires adventitielles sont souvent organisées en VALT (vascular associated lymphoid tissue) [14]. Ces structures pourraient être des sites fonctionnels d’activation de lymphocytes B résidents pathogènes de manière similaire à ce qui est observé dans les organes cibles des maladies auto-immunes [
15]. Le déclenchement de la réponse immune adaptative locale semble être indépendant des lésions de l’intima [
16]. |
Rôle des lymphocytes B résidents Doran et al. ont réussi, en modulant l’expression du récepteur de chimiokine CCR6, à empêcher la localisation artérielle des lymphocytes B chez les souris Apoe
-/-
, ce qui a accéléré et aggravé l’athérosclérose [16]. Ils ont ainsi démontré un rôle protecteur des lymphocytes B adventitiels, indépendant de leur production d’anticorps. Cette déplétion lymphocytaire entraîne aussi une diminution du nombre de macrophages résidents, peut-être via le déficit en facteurs de recrutement tissulaire qu’elle entraîne [
17]. De manière surprenante, l’inhibition de l’athérosclérose consécutive au transfert de lymphocytes B spléniques s’accompagne de la localisation, dans la paroi aortique, de lymphocytes majoritairement de type B2 [17]. Ce résultat semble contredire l’effet pathogène observé par Kyaw et al. [11]. Ces divergences peuvent être attribuées à plusieurs paramètres : génotype des souris, nombre de cellules transférées, régime alimentaire et stade de l’athérosclérose au moment de l’étude (les lymphocytes B présents dans les parois avant le déclenchement de la maladie préviendraient la formation des lésions [17]). Les modèles animaux d’athérosclérose (Tableau I) montrent que l’effet des lymphocytes B résidents dépend du contexte et de l’environnement tissulaires, qui peuvent altérer leur biologie. Plusieurs questions se posent : au sein de la paroi artérielle, la classification des lymphocytes en sous-populations B1 ou B2 suffit-elle à prédire leur rôle dans la maladie ? Ces lymphocytes B ont-ils des propriétés variables en fonction de l’évolution des lésions ? Les lymphocytes B2 pariétaux ont-ils la capacité de se différencier en Breg ou de sécréter des Ig protectrices ? En pathologie humaine, des considérations éthiques et pratiques limitent souvent l’accès aux lésions artérielles à celles de patients âgés à des stades avancés de leur maladie. Nous avons alors pu observer que les lymphocytes B des parois carotidiennes constituent une population cellulaire quantitativement importante (jusqu’à 70 % des éléments nucléés dans l’adventice artérielle) [
18]. Il existe, chez tous les malades, une synthèse locale d’IgA qu’accompagne celle d’IgG dans la majorité des cas. La présence d’IgM est plus rarement détectée (Figure 2). Les anticorps produits in situ pourraient contribuer aux dépôts d’Ig présents dans les parois artérielles pathologiques [18]. De nombreuses données expérimentales indiquent que les lymphocytes B résidents ont poursuivi leur maturation localement sous la pression de l’antigène. Ils expriment la cytidine déaminase AID (activation-induced cytidine deaminase), une enzyme clé catalysant les mutations somatiques et la commutation de classe des Ig. L’analyse des régions variables des Ig produites localement confirme cette observation en révélant une fréquence très élevée de mutations adaptatives1 [
33]. Au voisinage des lésions, le répertoire des immunoglobulines synthétisées est restreint, ce qui indique que les anticorps ont été sélectionnés pour ne reconnaître qu’un nombre limité de spécificités antigéniques (un lymphocyte B qui n’est pas stimulé de manière continue par l’antigène auquel il est adapté meurt rapidement et ne peut donc survivre durablement dans un tissu) [18,
19]. Dans plusieurs cas, l’évolution convergente de différents clones lymphocytaires, appartenant à des individus différents, aboutit à la formation d’Ig possédant des régions de complémentarité identiques avec les antigènes. Cette redondance est en accord avec la restriction des répertoires. La nature des antigènes locaux reste à ce jour inconnue. De manière intéressante, la majorité des lymphocytes B résidents sont des cellules très différenciées, de type B2, ayant perdu le marqueur CD20 et secrétant des cytokines de type inflammatoire (IL-6, GM-CSF [granulocyte macrophage-colony stimulating factor] et TNF-α [tumor necrosis factor-α]) (Figure 1). En revanche, ils ne synthétisent pas de cytokines anti-inflammatoires (IL-10, TGF-β [transforming growth factor-β]) dans les lésions. Il existe cependant une présence minoritaire et occasionnelle de lymphocytes B1 producteurs d’IgM naturelles (Figure 2).
 | Figure 2.
Rôles potentiels des lymphocytes B résidents dans l’athérosclérose humaine. Les lymphocytes B1 minoritaires inhibent l’athérosclérose en sécrétant des IgM protectrices et potentiellement de l’IL-10. Les fonctions athérogènes des lymphocytes B2 sont dépendantes de l’activation et de la prolifération des cellules T, ainsi que de la production d’Ig délétères (IgG, IgA). Les lymphocytes B2 pourraient également contribuer à la physiopathologie de l’athérosclérose en sécrétant des cytokines (IL-6, GM-CSF, TNF-α) qui réguleraient l’activité et l’infiltration des cellules immunitaires, en particulier des macrophages. MHC-II : major histocompatibility complex class-II. |
|
Réponse lymphocytaire B « innée » Les lymphocytes B1 inhibent la progression de l’athérosclérose chez la souris. Cet effet favorable semble lié à la production systémique d’Ig naturelles (le plus souvent IgM), car le transfert de lymphocytes B1, dont la production d’Ig est inhibée, est inefficace [20], confirmant ainsi le rôle protecteur des IgM établi lors d’études antérieures : la taille des lésions vasculaires chez les souris Ldlr
-/-
IgM
-/-
était en effet augmentée [21]. Les IgM naturelles ciblent souvent les lipoprotéines oxydées ou les cellules apoptotiques, contribuant ainsi à leur clairance. De plus, l’interaction IgM-LDLox inhibe l’internalisation des lipoprotéines, la formation de cellules spumeuses ainsi que l’activation endothéliale [20]. Le taux de ces IgM a été inversement corrélé à la sévérité des lésions [20]. L’élimination préférentielle des lymphocytes B1 par splénectomie semble être le déterminant de l’effet adverse de cette dernière sur l’évolution des lésions vasculaires. L’utilisation de souris splénectomisées pose néanmoins un problème méthodologique, car les lymphocytes B1 requièrent une maturation splénique pour une production optimum d’IgM [
22]. L’existence d’autres types d’anticorps protecteurs, bien que vraisemblable, reste encore à démontrer. Outre leur production d’anticorps naturels, les lymphocytes B1 sont une source majeure d’IL-10. Cette cytokine diminue l’athérosclérose en inhibant la présentation antigénique et la production de médiateurs inflammatoires [20]. Cependant, la présence dans les lésions, ou dans leur voisinage, de lymphocytes B1, ainsi que leur production d’IL-10, n’ont pas encore été explorées. Il est possible que leur rôle soit minoritaire et/ou transitoire au cours du développement de la maladie. L’étude des lymphocytes B1 humains souffre de l’absence de marqueurs spécifiques [12]. Une sous-population de lymphocytes B CD20+CD27+CD43+CD70-, récemment identifiée, pourrait cependant correspondre fonctionnellement aux lymphocytes B1 de la souris (→) [20, 32].
(→) Voir la Nouvelle de J.D. Bouaziz et al., m/s 2014, n° 8–9, page 721
|
Réponse lymphocytaire B adaptative Dans l’athérosclérose expérimentale de la souris, l’administration systémique d’anticorps anti-CD20 permet d’éliminer sélectivement les lymphocytes B2 sans affecter les lymphocytes B1. De manière concomitante, une réduction de la taille des lésions vasculaires est observée chez les animaux athéroscléreux traités [10, 11]. Dans ce contexte, la déplétion des lymphocytes B a des effets directs en diminuant les concentrations sérique et locale d’IgG sans modifier celles des IgM. Elle réduit aussi le nombre des lymphocytes B capables de capter les antigènes lipidiques des LDL et de les présenter aux lymphocytes NKT (lymphocytes natural killer T) [
23]. L’élimination des lymphocytes B2 agit aussi de manière indirecte en modifiant la réponse inflammatoire locale et systémique des lymphocytes T : elle s’accompagne d’une diminution de l’activité et de la sécrétion d’IFN-γ (interféron γ) des cellules T CD4+ spléniques, ainsi que d’une diminution du nombre de ces lymphocytes dans la plaque athéromateuse. En revanche, une augmentation de la production de la cytokine pro-inflammatoire IL-17 a été détectée chez les souris traitées par l’anticorps anti-CD20 (Figure 3) [10]. En résumé, ces observations indiquent que les lymphocytes B2 aggravent la maladie par voie humorale autant que cellulaire [10, 11]. Des preuves supplémentaires du rôle pathogène des lymphocytes B2 ont été apportées par l’invalidation du gène codant pour le récepteur de la cytokine BAFF (B-cell activating factor belonging to the TNF family), qui entraîne une inhibition sélective de la maturation lymphocytaire [20].
 | Figure 3.
Fonctions attribuees aux lymphocytes B systemiques dans l’atherosclerose. Outre leur production d’IgG, les lymphocytes B2 aggravent l’atherosclerose en induisant une deviation de la reponse immunitaire de Th17 vers Th1. Ces lymphocytes B2 peuvent egalement presenter les antigenes lipidiques aux cellules NKT. Par ailleurs, les lymphocytes B1, stimules par les cytokines Th2, inhibent l’atherosclerose en secretant des anticorps naturels anti-LDlox. la depletion des lymphocytes B2, ainsi que la stimulation des lymphocytes B1 par l’immunisation, sont des therapies potentielles d’inhibition de l’atherosclerose. |
Des études épidémiologiques ont révélé des effets opposés résultant de la production d’anticorps par les lymphocytes B2. Palinski et al. ont trouvé une corrélation positive entre les taux d’IgG anti-LDLox et l’évolution de la maladie des souris Ldlr
-/- [
24], alors que les IgG contre les peptides de l’ApoB-100 sont protecteurs chez les souris Apoe
-/-
[12]. À l’inverse du taux d’IgM, le taux d’IgG anti-LDLox est positivement corrélée à la sévérité de l’athérosclérose humaine [12]. Les complexes immuns humains IgG-anti-LDLox déclenchent la formation des cellules spumeuses et l’accumulation intracellulaire de cholestérol [
25]. Nicoletti et al. ont démontré que la suppression de la réponse IgG anti-LDLox par induction d’une tolérance immunitaire réduit la progression de l’athérosclérose des souris Apoe
-/-
[
26]. Par ailleurs, l’immunisation de souris Apoe
-/-
avec des LDLox augmente le taux d’IgG anti-LDLox, mais diminue cependant la taille des lésions et le taux de cholestérol [
27]. Il est intéressant d’observer des effets similaires après immunisation ou induction de tolérance. Ce paradoxe peut être attribué à la pluralité des mécanismes activés par ces traitements, ainsi qu’à la diversité des isotypes d’Ig recrutés. La variété d’action des IgG est en partie due aux types de récepteurs cellulaires d’Ig (FcγR) activés par les protocoles expérimentaux : la liaison au FcγRI est pathogène, alors que la signalisation par le FcγRIIb est protectrice [
28,
29]. |
Dans l’athérosclérose expérimentale, les populations lymphocytaires B1 et B2 ont des propriétés opposées et leur influence sur l’évolution des lésions artérielles découle en partie de l’équilibre entre leurs effets pro- ou anti-athérogènes (Figure 3). Cependant, cette résultante peut aussi varier au cours de la progression de la maladie et être modifiée par les différentes interactions cellulaires mises en jeu. La déplétion des lymphocytes B met en lumière leur contribution à la physiopathologie de l’athérosclérose, indépendamment de leur production d’anticorps protecteurs ou pathogènes. Les lymphocytes B participent à l’immunité à médiation cellulaire en modulant la réponse T par des processus encore mal connus, mais dont un des mécanismes probable est la sécrétion de cytokines qu’il serait utile de caractériser plus en détails afin de tenter de la réguler. La présentation d’antigènes par les lymphocytes B, ainsi que leur production de médiateurs de l’inflammation, sont d’autres facteurs importants de leur participation au processus athéromateux. Les cellules immunitaires sont présentes dans l’artère normale et pathologique. Leurs caractéristiques fonctionnelles se modifient avec le temps et/ou l’état évolutif de l’athérosclérose [
30]. La protection apportée par les lymphocytes B résidents murins aux stades précoces de la maladie contraste avec la production locale d’Ig et de cytokines pro-inflammatoires par les lymphocytes B adventitiels humains aux stades tardifs. Une étude chronologique des sous-populations de lymphocytes B dans la paroi artérielle permettrait de les caractériser et d’explorer leur cinétique d’infiltration afin de déterminer à quels stades il serait utile de limiter l’expansion de populations pathogènes ou d’amplifier l’action de populations régulatrices. La modulation de l’immunité locale est une voie prometteuse pour inhiber l’inflammation tissulaire sans affecter la réponse immune globale. Il est toujours délicat de transposer directement les données recueillies dans les modèles animaux aux situations pathologiques humaines, ainsi que d’envisager des protocoles thérapeutiques rigoureusement parallèles. Les traitements ablatifs par anticorps anti-CD20 s’adressent actuellement indifféremment à de nombreux types de lymphocytes B. De plus, bien que cette approche soit utilisée au cours de différentes maladies humaines [34] et qu’elle soit efficace sur l’athérosclérose murine, elle ne représente actuellement chez l’homme qu’une potentialité thérapeutique. La prise en compte d’autres caractéristiques fonctionnelles des lymphocytes B permettrait d’accroître les possibilités d’intervention thérapeutique en ciblant sélectivement les lymphocytes délétères (antagonistes de BAFF réduisant l’activation des lymphocytes B2) ou en stimulant les cellules protectrices (production d’anticorps naturels protecteurs s’appuyant sur des protocoles d’immunisation spécifiques contre les molécules pro-athérogènes). |
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
|
Footnotes |
1.
Caligiuri
G
. Rôle de l’immunité dans l’athérosclérose et dans les syndromes coronariens aigus . Med Sci (Paris).
2004; ; 20 : :175.–181. 2.
Anderson
GF
,
Chu
E
. Expanding priorities. Confronting chronic disease in countries with low income . N Engl J Med.
2007; ; 356 : :209.–211. 3.
Emmerich
J
. L’athérosclérose .
Collection Pathologie Science. , Paris: : John Libbey Eurotext; , 2000. 4.
Chisolm
GM
,
Steinberg
D
. The oxidative modification hypothesis of atherogenesis: an overview . Free Radic Biol Med.
2000; ; 28 : :1815.–1826. 5.
Campbell
KA
,
Lipinski
MJ
,
Doran
AC
, et al.
Lymphocytes and the adventitial immune response in atherosclerosis . Circ Res.
2012; ; 110 : :889.–900. 6.
Hamze
M
,
Desmetz
C
,
Guglielmi
P
. B cell-derived cytokines in disease . Eur Cytokine Netw.
2013; ; 24 : :20.–26. 7.
De Jager
SCA
,
Kuiper
J
. Vaccination strategies in atherosclerosis . Thromb Haemost.
2011; ; 106 : :796.–803. 8.
Caligiuri
G
,
Nicoletti
A
,
Poirier
B
,
Hansson
GK
. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice . J Clin Invest.
2002; ; 109 : :745.–753. 9.
Major
AS
,
Fazio
S
,
Linton
MF
. B-Lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice . Arterioscler Thromb Vasc Biol.
2002; ; 22 : :1892.–1898. 10.
Ait-Oufella
H
,
Herbin
O
,
Bouaziz
JD
, et al.
B cell depletion reduces the development of atherosclerosis in mice . J Exp Med.
2010; ; 207 : :1579.–1587. 11.
Kyaw
T
,
Tay
C
,
Khan
A
, et al.
Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis . J Immunol.
2010; ; 185 : :4410.–4419. 12.
Ponnuswamy
P
,
Van Vre
EA
,
Mallat
Z
,
Tedgui
A
. Humoral and cellular immune responses in atherosclerosis: spotlight on B- and T-cells . Vascul Pharmacol.
2012; ; 56 : :193.–203. 13.
Mironova
M
,
Virella
G
,
Lopes-Virella
MF
. Isolation and characterization of human antioxidized LDL autoantibodies . Arterioscler Thromb Vasc Biol.
1996; ; 16 : :222.–229. 14.
Kyaw
T
,
Tipping
P
,
Toh
BH
,
Bobik
A
. Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis . Curr Opin Lipidol.
2011; ; 22 : :373.–379. 15.
Aloisi
F
,
Pujol-Borrell
R
. Lymphoid neogenesis in chronic inflammatory diseases . Nat Rev Immunol.
2006; ; 6 : :205.–217. 16.
Lusis
AJ
. Atherosclerosis . Nature.
2000; ; 407 : :233.–241. 17.
Doran
AC
,
Lipinski
MJ
,
Oldham
SN
, et al.
B-cell aortic homing and atheroprotection depend on Id3 . Circ Res.
2012; ; 110 : :e1.–e12. 18.
Hamze
M
,
Desmetz
C
,
Berthe
ML
, et al.
Characterization of resident B cells of vascular walls in human atherosclerotic patients . J Immunol.
2013; ; 191 : :3006.–3016. 19.
Burioni
R
,
Canducci
F
,
Saita
D
, et al.
Antigen-driven evolution of B lymphocytes in coronary atherosclerotic plaques . J Immunol.
2009; ; 183 : :2537.–2544. 20.
Kyaw
T
,
Tipping
P
,
Bobik
A
,
Toh
BH
. Protective role of natural IgM-producing B1a cells in atherosclerosis . Trends Cardiovasc Med.
2012; ; 22 : :48.–53. 21.
Lewis
MJ
,
Malik
TH
,
Ehrenstein
MR
, et al.
Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient lice . Circulation.
2009; ; 120 : :417.–426. 22.
Perry
HM
,
Bender
TP
,
McNamara
CA
. B cell subsets in atherosclerosis . Front Immunol.
2012; ; 3 : :373.. 23.
Allan
LL
,
Hoefl
K
,
Zheng
DJ
, et al.
Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells . Blood.
2009; ; 114 : :2411.–2416. 24.
Palinski
W
,
Tangirala
RK
,
Miller
E
, et al.
Increased autoantibody titers against epitopes of oxidized LDL in LDL receptor-deficient mice with increased atherosclerosis . Arterioscler Thromb Vasc Biol.
1995; ; 15 : :1569.–1576. 25.
Klimov
AN
,
Denisenko
AD
,
Vinogradov
AG
, et al.
Accumulation of cholesteryl esters in macrophages incubated with human lipoprotein-antibody autoimmune complex . Atherosclerosis.
1988; ; 74 : :41.–46. 26.
Nicoletti
A
,
Paulsson
G
,
Caligiuri
G
, et al.
Induction of neonatal tolerance to oxidized lipoprotein reduces atherosclerosis tn ApoE knockout mice . Mol Med.
2000; ; 6 : :283.–290. 27.
Zhou
XH
,
Caligiuri
G
,
Hamsten
A
, et al.
LDL immunization induces T-cell-dependent antibody formation and protection against atherosclerosis . Arterioscler Thromb Vasc Biol.
2001; ; 21 : :108.–114. 28.
Hernandez-Vargas
P
,
Ortiz-Munoz
G
,
Lopez-Franco
O
, et al.
Fc gamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice . Circ Res.
2006; ; 99 : :1188.–1196. 29.
Mendez-Fernandez
YV
,
Stevenson
BG
,
Diehl
CJ
, et al.
The inhibitory Fc gamma RIIb modulates the inflammatory response and influences atherosclerosis in male apoE-/- mice . Atherosclerosis.
2011; ; 214 : :73.–80. 30.
Galkina
E
,
Kadl
A
,
Sanders
J
, et al.
Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent . J Exp Med.
2006; ; 203 : :1273.–1282. 31.
Varthaman
A
,
Khallou-Laschet
J
,
Thaunat
O
, et al.
L’athérogenèse : une maladie dysimmunitaire . Med Sci (Paris).
2008; ; 24 : :169.–176. 32.
Bouaziz
JD
,
de Masson
A
,
Le Buanec
H
, et al.
Lymphocytes B régulateurs : état des connaissances . Med Sci (Paris).
2014; ; 30 : :721.–724. 33.
Catalan
N
,
Imai
K
,
Revy
P
, et al.
Deux ans après, l’activation induced cytidine deaminase n’a pas livré tous ses secrets . Med Sci (Paris).
2003; ; 19 : :139.–141. 34.
Cartron
G
,
Rossi
JF
. Anticorps monoclonaux thérapeutiques en oncohématologie . Med Sci (Paris).
2009; ; 25 : :1085.–1089. |




