 |
| |||
Med Sci (Paris). 2014 October; 30(10): 889–895. Published online 2014 October 14. doi: 10.1051/medsci/20143010016.Le déficit en alpha-1-antitrypsine 1Institut de biochimie et génétique cellulaires, CNRS UMR 5095, université de Bordeaux, 1, rue Camille Saint-Saëns, 33077Bordeaux, France | |||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||
L’alpha-1-antitrypsine (α1AT) est une glycoprotéine d’environ 52 kDa appartenant à la superfamille des serpines (serine protease inhibitors) ; c’est l’antiprotéase sérique la plus abondante [ 1, 2]. Elle est produite principalement dans le foie par les hépatocytes, bien que d’autres cellules telles que les monocytes, les macrophages alvéolaires, les neutrophiles, les cellules bronchiques et alvéolaires (pneumocytes de type II) expriment également l’α1AT à un taux moindre [ 3– 6]. Le foie adulte sécrète plus d’un gramme d’α1AT par jour [ 7] dans la circulation sanguine où elle inhibe des protéases pro-inflammatoires telles que l’élastase, la protéinase-3 ainsi que de nombreuses cathepsines [ 8]. Ces protéases sont produites et sécrétées par des cellules pro-inflammatoires, telles que les polynucléaires neutrophiles, en réponse à l’inhalation d’agents pathogènes et/ou toxiques pro-inflammatoires (fumée de cigarette, etc.). Dans ce cas, les composants de la réponse inflammatoire sont étroitement contrôlés par différents systèmes, dont les antiprotéases comme l’α1AT, afin d’éviter une réponse qui pourrait devenir délétère pour les poumons. Par exemple, l’α1AT protège l’élastine (composant majeur de la matrice extracellulaire des alvéoles) d’une dégradation par les protéases (comme l’élastase). La production d’α1AT par le foie est donc cruciale pour maintenir l’équilibre entre protéases et antiprotéases [7, 9, 10]. Le déficit en α1AT est caractérisé par une diminution du taux sérique d’α1AT et donc de sa fonction d’inhibiteur de protéases. Les patients déficitaires en α1AT développent alors de graves problèmes respiratoires, tels que l’emphysème panlobulaire [ 11]. Dans les cas les plus sévères de la pathologie, c’est-à-dire chez les porteurs de la mutation homozygote ZZ, outre des lésions emphysémateuses pulmonaires, des anomalies hépatiques peuvent être observées [11]. De façon générale, les symptômes respiratoires associés au déficit en α1AT apparaissent entre 20 et 30 ans et sont souvent ceux d’une bronchopathie chronique asthmatiforme [ 12, 13]. L’emphysème pulmonaire devient généralement symptomatique aux alentours de 40-50 ans et est largement influencé par le tabac [12, 13]. À l’inverse, les anomalies hépatiques et l’ictère cholestatique apparaissent plus tôt, parfois dès la naissance [12, 13]. Avant l’âge de 20 ans, 3 % des patients développent une cirrhose, et après 25 ans, 8 à 10 % des patients homozygotes ZZ présentent un bilan hépatique perturbé (anomalies hépatiques et cirrhose) [12, 13]. Il n’existe à ce jour aucun traitement étiologique pour soigner ce déficit. Seuls des traitements substitutifs sont disponibles. Ils consistent en l’inhalation de bronchodilatateur (formotérol ou salmétérol) et l’injection d’α1AT purifiée. Cette dernière option est très controversée et son efficacité réelle reste encore à démontrer [7, 10]. Cette revue a pour but de faire un état des lieux des connaissances actuelles concernant le déficit en α1AT et les différents progrès réalisés ces dernières années concernant le traitement de ce déficit. | |||||||||||||||||||||||||||||||||||||||||||||
Le déficit en α1AT est une maladie génétique, héréditaire, de transmission autosomique codominante. Le gène responsable de ce déficit, SERPINA1, est localisé sur le chromosome 14 (14q31-32.3). Les premiers à décrire le déficit en α1AT sont Laurell et Eriksson en 1963. Lors de l’examen de protéines sériques par électrophorèse, Laurell remarqua l’absence de la bande représentant l’α1AT dans cinq des 1 500 sérums analysés. Curieusement, trois de ces cinq patients présentaient un emphysème précoce [ 14]. En 1969, Sharp et al. ont associé le déficit en α1AT à des anomalies hépatiques, en mettant en évidence la diminution d’α1AT chez 10 enfants souffrant de cirrhose [ 15]. Comme cela a été décrit précédemment, les conséquences physiopathologiques du déficit en α1AT sont donc des lésions pulmonaires et des anomalies hépatiques dans les cas les plus graves. Une personne saine présente une concentration en α1AT circulante de 0,9 à 2 g/l ; une personne est considérée comme déficitaire en α1AT lorsque cette concentration est inférieure à 0,8 g/l. La prévalence du déficit en α1AT en Europe et en Amérique du Nord est d’environ une naissance sur 2 000 [11]. Ce déficit représente la cause génétique la plus commune d’anomalies du foie chez l’enfant [ 16]. Malgré cela, le déficit en α1AT est insuffisamment diagnostiqué, car les symptômes associés sont souvent reliés à des causes exogènes, comme le tabagisme. | |||||||||||||||||||||||||||||||||||||||||||||
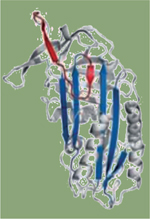
Plus de 90 mutations de l’α1AT ont été identifiées. Les mutations ponctuelles, appelées S et Z, sont les plus communément rencontrées (Tableau I). Le mutant Z est le plus sévère. Son nom provient de sa mobilité électrophorétique, particulièrement lente par comparaison à la forme sauvage ou à la forme mutante S pour slowly (Tableau I). Une mutation ponctuelle (la substitution d’une lysine par un glutamate en position 342 [E342K]) conduit au repliement incorrect de la protéine mutée Z (Tableau I) [9]. Comme tous les autres mutants, le variant Z n’est plus sécrété par les hépatocytes, ce qui entraîne une diminution d’α1AT dans le sérum et donc, à terme, à un emphysème pulmonaire. À l’inverse des autres mutants qui sont généralement dégradés par le protéasome, la mutation E342K induit la formation et l’accumulation de polymères (polymères Z) dans le réticulum endoplasmique (Figure 1). Cette accumulation peut, dans certains cas, être toxique pour la cellule, conduisant à la mort des hépatocytes et, par conséquent, à des anomalies hépatiques observés chez 8 % des patients homozygotes ZZ (Tableau I) [ 17].
Biologie cellulaire du mutant Z Bien que le variant Z ne soit pas correctement replié, il conserve sa capacité à inhiber les protéases [
18]. Par conséquent, la compréhension des mécanismes régissant la biogenèse et les fonctions de cette protéine est très importante. L’identification des partenaires protéiques de la protéine Z dans le réticulum endoplasmique pourra donc contribuer à la compréhension des mécanismes de rétention réticulaire de cette protéine et, ainsi, ouvrir de nouvelles opportunités thérapeutiques.La théorie actuelle, basée sur des données expérimentales, suggère qu’il existerait trois catégories distinctes de protéines Z au sein de la cellule [ 19] : (1) l’une correspondant à la protéine sécrétée ; (2) une seconde à la protéine agrégée, non soluble ; et, enfin, (3) une troisième à la protéine soluble destinée à la dégradation par le protéasome. Cette dernière catégorie représente la majeure partie des formes intracellulaires du variant Z (> 85 %). Néanmoins, avant d’être réparti dans ces différentes catégories, le variant Z interagit avec de nombreuses protéines dans le réticulum endoplasmique, qui déterminent alors son devenir dans la cellule (dégradation/rétention/sécrétion). Après sa traduction, la protéine Z naissante acquiert trois extensions N-glycanes, puis elle est prise en charge par un système de contrôle de qualité des glycoprotéines dans la lumière du réticulum endoplasmique et, en particulier, par la protéine majeure de ce système, la calnexine. Cette dernière joue un rôle essentiel dans les mécanismes de rétention du mutant Z dans le réticulum endoplasmique [19– 22]. La réduction de l’expression de calnexine est suffisante pour rétablir le trafic de ce mutant. De plus, l’utilisation de molécules telles que la kifunensine (inhibiteur des mannosidases I et II) ou la castanospermine (inhibiteur de glucosidases), qui affectent indirectement l’interaction du mutant Z avec la calnexine, permet de rétablir la sécrétion de ce variant et ainsi son activité antiprotéase [19, 23]. Enfin, des modifications post-traductionnelles affectant la calnexine, comme sa phosphorylation ou son acétylation, pourraient également jouer un rôle essentiel dans le rétablissement de la sécrétion du mutant Z [19, 20]. De même, l’interaction du variant Z avec d’autres chaperonnes comme BiP/GRP78 (binding immunoglobulin protein/glucose-regulated protein 78) ou GRP94 (glucose-regulated protein 94) pourrait aussi jouer un rôle dans son repliement [ 24]. Malgré l’absence de pont disulfure dans l’α1AT, le rôle de la voie d’oxydoréduction semble être importante dans la biologie du variant Z. En effet, la protéine Z interagit avec PDI (protéine disulfide isomérase), ce qui entraîne l’inhibition de l’activité de PDI, générant alors un stress oxydatif [ 25]. De plus, l’atténuation de l’expression d’une autre protéine intervenant dans la voie d’oxydoréduction, la protéine ERO1Lα (endoplasmic oxidoreductin-1-like protein α ; protéine contrôlant l’oxydation de PDI), peut également rétablir la sécrétion du mutant Z [19]. Récemment, il a été mis en évidence que la forme soluble du variant Z dégradée par le protéasome est prise en charge par la voie de dégradation des glycoprotéines GERAD (glycoprotein endoplasmic-reticulum-associated protein degradation) et, plus particulièrement, par les protéines EDEM3/OS9/SEL1L (ER-degradation-enhancing-mannosidase-like protein 3/osteosarcoma amplified 9/suppressor of lin-12-like protein 1). Ces protéines sont responsables de l’adressage du variant Z au protéasome. La diminution de l’expression de la protéine EDEM3 est suffisante pour rétablir la sécrétion du variant Z [19]. Enfin, la protéine ERGIC-53 (ER-Golgi intermediate compartment of 53-kDa) a été identifiée comme le récepteur impliqué dans le trafic de l’α1AT sauvage entre le réticulum endoplasmique et l’appareil de Golgi [ 26]. L’extinction de l’expression d’ERGIC-53 dans un modèle cellulaire ou murin induit une accumulation de la forme sauvage d’α1AT dans le réticulum endoplasmique des hépatocytes [ 27]. Polymères/agrégats : formation et conséquences Des études de cristallographie ont révélé que la protéine α1AT présente trois feuillet β (A-C) et une boucle mobile appelée RCL (reactive center loop) contenant le centre actif (Figure 1). La substitution d’un seul acide aminé en position 342 (mutation Z) conduit à l’ouverture du feuillet β-A, ce qui facilite alors l’insertion partielle de la boucle RCL d’une autre protéine Z (Figure 1). Cela conduit à la formation d’un dimère qui se propage en polymères (Figure 1) [
28]. D’autres mutants extrêmement rares d’α1AT (Siiyama, Mmalton ;
Tableau I
) peuvent également perturber la structure du feuillet β-A et conduire à la formation de polymères (Figure 1). Le foie étant le site majeur de production d’α1AT, la polymérisation du variant Z est donc prédominante dans le réticulum endoplasmique des hépatocytes. Les polymères sont alors retenus dans la cellule sous forme de corps d’inclusion. Ces derniers peuvent être colorés à l’acide périodique de Schiff (PAS) et résistent à la digestion par la diastase [
29].L’accumulation des polymères Z peut se révéler toxique pour les hépatocytes. Parmi les patients homozygotes ZZ, seuls 8 % développent des anomalies hépatiques [ 30]. Il est maintenant évident que des facteurs génétiques et environnementaux ont une profonde influence sur le phénotype hépatique. Il a été récemment mis en évidence que des variations génétiques (polymorphismes nucléotidiques) au sein du gène codant pour la protéine ERManI (ER mannosidase I) influenceraient l’âge d’apparition des anomalies hépatiques chez les patients homozygotes ZZ [ 31]. Toutefois, les mécanismes par lesquels les polymères Z induisent la mort des hépatocytes restent non élucidés. La cellule hépatique se protège partiellement des polymères Z en activant l’autophagie pour dégrader ces polymères accumulés dans le réticulum endoplasmique. Une altération de cette voie de dégradation des protéines pourrait d’ailleurs être importante dans l’apparition des symptômes hépatiques [ 32]. Il a également été démontré que des mécanismes indépendants de l’autophagie et du protéasome pouvaient contribuer à la dégradation intracellulaire du variant Z. Récemment, il a été mis en évidence que le variant Z pouvait être dégradé à partir du Golgi par le lysosome [ 33]. D’autres voies non caractérisées sont donc susceptibles d’être impliquées dans la dégradation du variant Z. L’accumulation du variant Z au sein du réticulum endoplasmique des hépatocytes n’induit pas le stress du réticulum endoplasmique et n’active pas l’UPR (unfolded protein response) [ 34]. De façon générale, cette réponse adaptative est induite par l’accumulation de protéines mal conformées dans le réticulum endoplasmique. Elle vise à rétablir l’homéostasie de ce compartiment via l’activation des trois protéines médiatrices de la réponse UPR : PERK (PKR-related endoplasmic reticulum kinase), ATF6 (activating transcription factor 6) et IRE1 (inositol requiring enzyme 1) [ 35]. Néanmoins, l’activation d’une des branches de l’UPR consécutive à la surexpression de la forme nucléaire d’ATF6 induit une diminution de l’accumulation du variant Z et de sa toxicité cellulaire [ 36]. Bien que les polymères Z n’induisent pas l’UPR dans les hépatocytes, ils activent cette réponse dans les monocytes de patients ZZ [ 37]. Ces résultats semblent indiquer l’existence d’une spécificité cellulaire d’activation de l’UPR. Cela pourrait être dû à une sensibilité différentielle à l’accumulation de protéines mal conformées dans le réticulum endoplasmique, ou à un taux de protéines chaperonnes différent selon le type cellulaire. Finalement, bien que le variant Z n’induise pas l’UPR dans les hépatocytes, l’accumulation de ce mutant peut activer la voie pro-inflammatoire dépendante de NF-B appelé EOR (ER overload response) [ 38]. Malgré l’accumulation majeure de ce mutant dans le réticulum endoplasmique, de faibles quantités de variant Z sont sécrétées sous forme polymérique [19, 39]. Les polymères Z sont par conséquent retrouvés dans l’environnement extracellulaire (circulation, lavages pulmonaires, liquides interstitiels). Ils y induisent une réaction pro-inflammatoire : d’une part, les polymères colocalisent avec les neutrophiles dans les alvéoles pulmonaires des patients [ 40] ; d’autre part, ils sont chémo-attractants in vitro pour les neutrophiles et favorisent leur adhésion et stimulent leur dégranulation [9]. Enfin, l’environnement peut jouer un rôle important dans le déficit en α1AT, en particulier dans la formation des polymères Z. La fumée de cigarette conduit à l’oxydation du centre actif de l’α1AT sauvage et augmente la polymérisation du mutant Z dans les poumons [ 41]. | |||||||||||||||||||||||||||||||||||||||||||||
Actuellement, seuls des traitements palliatifs sont utilisés chez les patients ayant un déficit en α1AT. Ils incluent l’inhalation de bronchodilatateurs ou le traitement substitutif par l’injection d’α1AT purifiée. Cet éventail thérapeutique réduit explique la nécessité de développer de nouvelles stratégies pharmacologiques pour traiter le déficit en α1AT. Récemment, deux nouveaux médicaments pouvant rétablir la sécrétion du variant Z et réduire la rétention des polymères Z au sein du réticulum endoplasmique ont été identifiés (Tableau II). Il s’agit de l’inhibiteur des HDAC (histone déacétylases), le SAHA (acide subéroylanilide hydroxamique), et de l’inducteur de l’autophagie, la carbamazépine (CBZ) (Tableau II) [19, 32]. Un traitement par le SAHA rétablit la sécrétion du variant Z. Cet effet est causé, entre autres, par l’inhibition de la protéine HDAC7 et par la réduction de l’interaction entre le variant Z et la calnexine. D’autres inhibiteurs d’HDAC peuvent aussi rétablir la sécrétion du variant Z, comme le Scriptaid ou la chaperonne chimique 4-PBA (phénylbutyrate de sodium) (Tableau II) [19, 42, 43]. L’inhibiteur Scriptaid n’est pas autorisé par l’ANSM (Agence nationale de sécurité du médicament et des produits de santé) et le 4-PBA s’est révélé inefficace lors d’essais cliniques. Toutefois, l’inhibiteur SAHA, approuvé pour le traitement du lymphome T cutané, induit des effets secondaires digestifs (nausées, vomissements, anorexie) et une asthénie [ 44]. Ces résultats montrent toutefois que les HDAC pourraient représenter des cibles thérapeutiques intéressantes pour le traitement des atteintes pulmonaires secondaires au déficit en α1AT.
La CBZ, quant à elle, active l’autophagie et conduit à une importante réduction de la quantité des polymères Z dans le réticulum endoplasmique des hépatocytes, ainsi qu’à une réduction de la fibrose hépatique causée par le variant Z dans un modèle murin [32]. La CBZ augmente également la dégradation du variant Z par le protéasome (Tableau II) [32]. Récemment, un criblage à partir de cellules souches pluripotentes induites (iPSC, induced pluripotent stem cell) issues de patients ZZ et un criblage à partir du modèle C. elegans du déficit en α1AT ont permis d’identifier le VPA (acide valproïque), le lithium et la fluphénazine comme des agents pouvant également réduire la rétention des polymères Z dans le réticulum endoplasmique (Tableau II) [45, 46]. Tout comme la CBZ, ces molécules induisent l’autophagie. Une autre stratégie pour traiter le déficit en α1AT est la thérapie génique. Quatre essais cliniques ont été réalisés. Cependant, ils se sont avérés peu efficaces, puisqu’ils n’ont permis d’augmenter que de 3 à 5 % la concentration en α1AT sérique, ce qui est très inférieur à la quantité nécessaire pour rétablir une concentration suffisante d’α1AT circulante [ 47]. Une autre solution thérapeutique pourrait être l’utilisation de peptides ou d’oligonucléotides, ou encore le « remaniement du génome ». Ainsi, récemment, l’utilisation des nucléases à doigt de zinc en combinaison avec la technologie de piggybac a permis de corriger la mutation Z E342K dans des cellules souches pluripotentes induites (iPSC). Cette « correction génétique » permet alors de rétablir la structure et la fonction de l’α1AT in vitro et in vivo [ 48]. Toutefois, cette approche introduit également des mutations dans le génome qui pourraient s’avérer carcinogènes. Une autre option serait l’utilisation d’oligonucléotides antisens dirigés contre l’α1AT. Il a été montré que l’utilisation de cette stratégie réduit significativement l’accumulation du mutant Z et la fibrose hépatique dans un modèle murin du déficit en α1AT [ 49]. Enfin, l’utilisation d’un peptide ciblant le centre actif du variant Z a conduit à l’augmentation significative de la sécrétion d’une forme active du mutant Z et à la diminution de la formation des polymères [ 50]. | |||||||||||||||||||||||||||||||||||||||||||||
Le déficit en α1AT représente une susceptibilité génétique insuffisamment diagnostiquée qui, malgré tout, touche approximativement 1 individu sur 2 0001. La mutation Z est responsable de la forme la plus sévère de ce déficit. Cette mutation induit des lésions pulmonaires (emphysème) et hépatiques (cirrhose). Il n’existe à l’heure actuelle aucun traitement efficace contre le déficit en α1AT. Une meilleure compréhension des mécanismes régissant la biogenèse et les fonctions du variant Z amènera donc à l’identification de cibles thérapeutiques et de biomarqueurs. À titre d’exemple, des composés (SAHA, CBZ), utilisés dans des modèles cellulaires et/ou murins, ont permis de rétablir la sécrétion du variant Z, et de diminuer la quantité de ses polymères. La mise en évidence de ces composés, ainsi que la compréhension de leur mode d’action, apportent de réels espoirs dans le traitement du déficit en α1AT. | |||||||||||||||||||||||||||||||||||||||||||||
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. | |||||||||||||||||||||||||||||||||||||||||||||
Ce travail a été réalisé avec le soutien de la Fondation pour la recherche médicale (SPF 20130526734) et de l’Association française des déficitaires en alpha-1-antitrypsine. Je remercie également les membres du laboratoire de Christophe Cullin pour leur précieux conseils. | |||||||||||||||||||||||||||||||||||||||||||||
1
Association française des déficitaires en alpha-1-antitrypsine : http://www.alpha1-france.org/
| |||||||||||||||||||||||||||||||||||||||||||||
1.
Hunt
LT
,
Dayhoff
MO
. A surprising new protein superfamily containing ovalbumin, antithrombin-III, and alpha 1-proteinase inhibitor . Biochem Biophys Res Commun.
1980; ; 95 : :864.–871. 2.
Silverman
GA
,
Bird
PI
,
Carrell
RW
, et al.
The serpins are an expanding superfamily of structurally similar but functionally diverse proteins Evolution, mechanism of inhibition, novel functions, and a revised nomenclature . J Biol Chem.
2001; ; 276 : :33293.–33296. 3.
Cichy
J
,
Potempa
J
,
Travis
J
. Biosynthesis of alpha1-proteinase inhibitor by human lung-derived epithelial cells . J Biol Chem.
1997; ; 272 : :8250.–8255. 4.
Venembre
P
,
Boutten
A
,
Seta
N
, et al.
Secretion of alpha 1-antitrypsin by alveolar epithelial cells . FEBS Lett.
1994; ; 346 : :171.–174. 5.
Paakko
P
,
Kirby
M
,
du Bois
RM
, et al.
Activated neutrophils secrete stored alpha 1-antitrypsin . Am J Respir Crit Care Med.
1996; ; 154 : :1829.–1833. 6.
Mornex
JF
,
Chytil-Weir
A
,
Martinet
Y
, et al.
Expression of the alpha-1-antitrypsin gene in mononuclear phagocytes of normal and alpha-1-antitrypsin-deficient individuals . J Clin Invest.
1986; ; 77 : :1952.–1961. 7.
Bouchecareilh
M
,
Balch
WE
. Proteostasis, an emerging therapeutic paradigm for managing inflammatory airway stress disease . Curr Mol Med.
2012; ; 12 : :815.–826. 8.
Baugh
RJ
,
Travis
J
. Human leukocyte granule elastase: rapid isolation and characterization . Biochemistry.
1976; ; 15 : :836.–841. 9.
Gooptu
B
,
Ekeowa
UI
,
Lomas
DA
. Mechanisms of emphysema in alpha1-antitrypsin deficiency: molecular and cellular insights . Eur Respir J.
2009; ; 34 : :475.–488. 10.
Bouchecareilh
M
,
Balch
WE
. Proteostasis: a new therapeutic paradigm for pulmonary disease . Proc Am Thorac Soc.
2011; ; 8 : :189.–195. 11.
Stoller
JK
,
Aboussouan
LS
. A review of alpha1-antitrypsin deficiency . Am J Respir Crit Care Med.
2012; ; 185 : :246.–259. 12.
Gooptu
B
,
Dickens
JA
,
Lomas
DA
. The molecular and cellular pathology of alpha1-antitrypsin deficiency . Trends Mol Med.
2014; ; 20 : :116.–127. 13.
Ghouse
R
,
Chu
A
,
Wang
Y
,
Perlmutter
DH
. Mysteries of alpha1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease . Dis Model Mech.
2014; ; 7 : :411.–419. 14.
Laurell
CB
,
Eriksson
S
. The electrophoretic alpha-1-globulin pattern of serum in alpha-1-antitrypsin deficiency . Scand J Clin Lab Invest.
1963; ; 15 : :132.–140. 15.
Sharp
HL
,
Bridges
RA
,
Krivit
W
,
Freier
EF
. Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder . J Lab Clin Med.
1969; ; 73 : :934.–939. 16.
Perlmutter
DH
. Liver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injury . J Clin Invest.
2002; ; 110 : :1579.–1583. 17.
Perlmutter
DH
. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates . Annu Rev Med.
2011; ; 62 : :333.–345. 18.
Ogushi
F
,
Fells
GA
,
Hubbard
RC
, et al.
Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase . J Clin Invest.
1987; ; 80 : :1366.–1374. 19.
Bouchecareilh
M
,
Hutt
DM
,
Szajner
P
, et al.
Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency . J Biol Chem.
2012; ; 287 : :38265.–38278. 20.
Cameron
PH
,
Chevet
E
,
Pluquet
O
, et al.
Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway . J Biol Chem.
2009; ; 284 : :34570.–34579. 21.
Granell
S
,
Baldini
G
,
Mohammad
S
, et al.
Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function . Mol Biol Cell.
2008; ; 19 : :572.–586. 22.
Qu
D
,
Teckman
JH
,
Omura
S
,
Perlmutter
DH
. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity . J Biol Chem.
1996; ; 271 : :22791.–22795. 23.
Marcus
NY
,
Perlmutter
DH
. Glucosidase and mannosidase inhibitors mediate increased secretion of mutant alpha1 antitrypsin Z . J Biol Chem.
2000; ; 275 : :1987.–1992. 24.
Schmidt
BZ
,
Perlmutter
DH
. Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum . Am J Physiol Gastrointest Liver Physiol.
2005; ; 289 : :G444.–G455. 25.
Papp
E
,
Szaraz
P
,
Korcsmaros
T
,
Csermely
P
. Changes of endoplasmic reticulum chaperone complexes, redox state, and impaired protein disulfide reductase activity in misfolding alpha1-antitrypsin transgenic mice . FASEB J.
2006; ; 20 : :1018.–1020. 26.
Nyfeler
B
,
Reiterer
V
,
Wendeler
MW
, et al.
Identification of ERGIC-53 as an intracellular transport receptor of alpha1-antitrypsin . J Cell Biol.
2008; ; 180 : :705.–712. 27.
Zhang
B
,
Zheng
C
,
Zhu
M
, et al.
Mice deficient in LMAN1 exhibit FV and FVIII deficiencies and liver accumulation of alpha1-antitrypsin . Blood.
2011; ; 118 : :3384.–3391. 28.
Crowther
DC
,
Belorgey
D
,
Miranda
E
, et al.
Practical genetics: alpha-1-antitrypsin deficiency and the serpinopathies . Eur J Hum Genet.
2004; ; 12 : :167.–172. 29.
Marciniak
SJ
,
Lomas
DA
. Alpha1-antitrypsin deficiency and autophagy . N Engl J Med.
2010; ; 363 : :1863.–1864. 30.
Piitulainen
E
,
Carlson
J
,
Ohlsson
K
,
Sveger
T
. Alpha1-antitrypsin deficiency in 26-year-old subjects: lung, liver, and protease/protease inhibitor studies . Chest.
2005; ; 128 : :2076.–2081. 31.
Pan
S
,
Huang
L
,
McPherson
J
, et al.
Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency . Hepatology.
2009; ; 50 : :275.–281. 32.
Hidvegi
T
,
Ewing
M
,
Hale
P
, et al.
An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis . Science.
2010; ; 329 : :229.–232. 33.
Gelling
CL
,
Dawes
IW
,
Perlmutter
DH
, et al.
The endosomal protein-sorting receptor sortilin has a role in trafficking alpha-1 antitrypsin . Genetics.
2012; ; 192 : :889.–903. 34.
Hidvegi
T
,
Schmidt
BZ
,
Hale
P
,
Perlmutter
DH
. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response . J Biol Chem.
2005; ; 280 : :39002.–39015. 35.
Bouchecareilh
M
,
Chevet
E
. Stress du réticulum endoplasmique : une réponse pour éviter le pIRE . Med Sci (Paris).
2009; ; 25 : :281.–287. 36.
Smith
SE
,
Granell
S
,
Salcedo-Sicilia
L
, et al.
Activating transcription factor 6 limits intracellular accumulation of mutant alpha1-antitrypsin Z and mitochondrial damage in hepatoma cells . J Biol Chem.
2011; ; 286 : :41563.–41577. 37.
Carroll
TP
,
Greene
CM
,
O’Connor
CA
, et al.
Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency . J Immunol.
2010; ; 184 : :4538.–4546. 38.
Lawless
MW
,
Greene
CM
,
Mulgrew
A
, et al.
Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency . J Immunol.
2004; ; 172 : :5722.–5726. 39.
Miranda
E
,
Perez
J
,
Ekeowa
UI
, et al.
A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency . Hepatology.
2010; ; 52 : :1078.–1088. 40.
Mahadeva
R
,
Atkinson
C
,
Li
Z
, et al.
Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo
. Am J Pathol.
2005; ; 166 : :377.–386. 41.
Alam
S
,
Li
Z
,
Janciauskiene
S
,
Mahadeva
R
. Oxidation of Z alpha1-antitrypsin by cigarette smoke induces polymerization: a novel mechanism of early-onset emphysema . Am J Respir Cell Mol Biol.
2011; ; 45 : :261.–269. 42.
Burrows
JA
,
Willis
LK
,
Perlmutter
DH
. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency . Proc Natl Acad Sci USA.
2000; ; 97 : :1796.–1801. 43.
Mendre
C
,
Mouillac
B
. Chaperons pharmacologiques : un espoir thérapeutique pour les pathologies conformationnelles . Med Sci (Paris).
2010; ; 26 : :627.–635. 44.
Kelly
WK
,
O’Connor
OA
,
Krug
LM
, et al.
Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer . J Clin Oncol.
2005; ; 23 : :3923.–3931. 45.
Choi
SM
,
Kim
Y
,
Shim
JS
, et al.
Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells . Hepatology.
2013; ; 57 : :2458.–2468. 46.
Li
J
,
Pak
SC
,
O’Reilly
LP
, et al.
Fluphenazine reduces proteotoxicity in C. elegans, mammalian models of alpha-1-antitrypsin deficiency . PLoS One.
2014; ; 9 : :e87260.. 47.
Flotte
TR
,
Mueller
C
. Gene therapy for alpha-1 antitrypsin deficiency . Hum Mol Genet.
2011; ; 20 : :R87.–R92. 48.
Yusa
K
,
Rashid
ST
,
Strick-Marchand
H
, et al.
Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells . Nature.
2011; ; 478 : :391.–394. 49.
Guo
S
,
Booten
SL
,
Aghajan
M
, et al.
Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin-related liver disease in mice . J Clin Invest.
2014; ; 124 : :251.–261. | |||||||||||||||||||||||||||||||||||||||||||||

