 |
| |||
Med Sci (Paris). 2016 March; 32(3): 274–280. Published online 2016 March 23. doi: 10.1051/medsci/20163203012.Réabsorption du sel et sécrétion du potassium par le néphron distal Vision nouvelle du rôle régulateur des kinases de la famille WNK 1Inserm UMR970, Paris Centre de Recherche cardiovasculaire, 56, rue Leblanc, 75015Paris, France 2Université Paris-Descartes, Sorbonne Paris Cité, Paris, France 3Université Paris-Diderot, Sorbonne Paris Cité, Paris, France Corresponding author. | ||||||
| ||||||
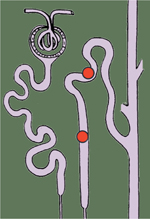
L’hypertension artérielle essentielle (HTA) est une maladie complexe, causée par une combinaison de facteurs génétiques et environnementaux. L’un des facteurs de risque les plus connus est l’exposition chronique à un régime trop riche en chlorure de sodium [1, 2] mais les mécanismes responsables de cette sensibilité de la pression artérielle au sel n’ont pas encore été complétement élucidés. Cependant, grâce à une modélisation mathématique complexe, A.C. Guyton proposait en 1972 qu’un défaut de transport ionique dans le néphron était impliqué dans toutes les formes d’HTA [3]. Une des stratégies d’étude utilisées pour identifier de nouveaux facteurs mis en jeu dans la régulation de la pression artérielle est l’analyse génétique de formes caricaturales, rares et mendéliennes d’hypertension artérielle. L’hypertension hyperkaliémique familiale (FHHt), également connue sous le nom de syndrome de Gordon ou pseudohypoaldostéronisme de type 2, est l’un de ces syndromes [4]. Les patients atteints de FHHt présentent une hypertension modérée, une hyperkaliémie1, et une acidose métabolique hyperchlorémique2,. L’ensemble de ces troubles est corrigé par une faible dose de diurétiques thiazidiques3 [33] (→), qui inhibent le co-transporteur Na+-Cl- NCC (Na+-Cl− cotransporter) exprimé dans le segment du néphron appelé tubule contourné distal (DCT) (Figure 1). Cette sensibilité aux diurétiques thiazidiques a laissé supposer que la FHHt avait pour cause une activation du co-transporteur NCC. Des mutations inactivatrices de NCC sont responsables du syndrome de Gitelman, un syndrome miroir de la FHHt (qui se caractérise entre autres par une alcalose hypokaliémique). Cependant, aucune mutation du gène codant NCC (SLC12A3 [solute carrier family 12, member 3]) n’a été identifiée chez les patients atteints de FHHt. De plus, les analyses de liaisons génétiques n’ont pas permis de mettre en évidence une association significative entre la FHHt et le bras court du chromosome 16, où est localisé le gène SLC12A3. (→) Voir la Nouvelle de D. Eladari et al., m/s n° 5, mai 2010, page 549
En 2001, les premières mutations responsables de la FHHt ont été identifiées [5]. Elles sont localisées dans les gènes qui codent deux membres de la famille des sérine thréonine kinases WNK (with no lysine [K]), WNK1 et WNK4, qui sont exprimées dans le rein. Le nom étrange de ces kinases est dû au remplacement de la lysine catalytique, présente dans le sous-domaine II de la majorité des protéine kinases connues, par une cystéine [6]. De nombreuses équipes ont étudié la régulation de NCC par les kinases WNK1 et WNK4 [34] (→). (→) Voir la Synthèse de J. Hadchouel et al., m/s n° 1, janvier 2005, page 55 Cependant, les résultats obtenus étaient souvent contradictoires et un modèle clair et unique expliquant le mécanisme de régulation de l’expression et de l’activité du cotransporteur NCC par les WNK n’a jamais pu être établi. En effet, en fonction des modèles utilisés, WNK4 et WNK1 présentaient, dans certaines études, un rôle activateur de NCC et, dans d’autres, un rôle inhibiteur (voir plus loin). Deux publications récentes de notre équipe ont permis de commencer à expliquer ces contradictions [7, 8]. La première démontre que les résultats contradictoires des études portant sur la régulation de NCC par WNK1 provenaient de l’utilisation d’un ADN complémentaire (ADNc) de WNK1 présentant une mutation inactivatrice [8]. La deuxième montre que les effets activateurs et inhibiteurs de WNK4 sur la régulation de NCC sont en fait présents au sein d’une même cellule et qu’ils sont modulés par la concentration intracellulaire de chlorure [7]. Dans cette revue, nous reviendrons sur ces récentes découvertes et nous proposerons un nouveau modèle pour expliquer le rôle que jouent WNK1 et WNK4 dans la régulation du transport de NaCl et de potassium dans le rein. | ||||||
WNK1, activateur de SPAK ou inhibiteur de WNK4 ? Les premières études portant sur la régulation de NCC par les WNK ont été menées in vitro en utilisant des œufs de xénope4 ou des cultures de lignées cellulaires. Dans ces modèles, la surexpression de WNK4 inhibe l’expression membranaire de NCC et donc son activité [9, 10]. Ce mode de régulation dépend de l’activité kinase de WNK4 puisqu’un mutant WNK4-kinase dead, qui ne présente plus d’activité kinase, n’est plus capable d’inhiber NCC.Les mécanismes par lesquels WNK1 régule NCC sont longtemps restés controversés. Deux isoformes de WNK1 ont été décrites [11] : L-WNK1 (long-WNK1), qui est ubiquitaire et possède une activité catalytique, et KS-WNK1 (kidney-specific-WNK1), qui est une forme tronquée de la protéine exprimée spécifiquement dans la partie distale du néphron et qui est dépourvue d’activité kinase. Dans la suite, nous nous intéresserons essentiellement à L-WNK1 qui est l’isoforme impliquée dans la FHHt [12]. Plusieurs études avaient montré que la surexpression de L-WNK1, seul, dans l’œuf de xénope ou les cellules rénales MDCK (Madin-Darby canine kidney) ne modifiait pas l’activité de NCC [10, 13]. Pourtant, L-WNK1 peut activer NCC par inhibition de WNK4. Parallèlement, des études biochimiques avaient montré que L-WNK1 pouvait activer NCC indépendamment de WNK4. En effet, L-WNK1 phosphoryle la kinase SPAK (Ste20-related proline-alanine rich kinase) qui, elle-même, phosphoryle et active NCC [14]. Deux voies de régulation de NCC par L-WNK1 étaient donc possibles. Afin de déterminer la ou les voies impliquées in vivo, nous avons utilisé un modèle de souris génétiquement modifiées. L’inactivation du gène L-WNK1 provoque la mort des embryons avant le 13e jour de développement [15]. Il est donc impossible d’étudier dans ce modèle la régulation de NCC par la kinase. Nous avons donc généré un modèle de surexpression de la protéine L-WNK1. Les mutations de WNK1 impliquées dans la FHHt sont de larges délétions de l’intron 1 du gène. Elles provoquent une surexpression de la protéine L-WNK1 dans les leucocytes des patients [5]. Nous avons éliminé le premier intron de WNK1 chez la souris (souris WNK1+/FHHt ) et avons ainsi obtenu un modèle présentant l’ensemble des phénotypes associés à la FHHt [12]. Cette délétion intronique entraîne une augmentation de l’expression de L-WNK1 dans le DCT et le tubule connecteur sans que l’expression de KS-WNK1 ne soit modifiée. Comme attendu, l’abondance et la phosphorylation de la protéine NCC sont augmentées. Nous avons également observé une augmentation de la forme phosphorylée de SPAK à la membrane apicale des cellules du DCT. Des travaux plus récents, utilisant le même modèle murin WNK1+/FHHt mais qui n’exprime pas WNK4, ont confirmé la capacité de L-WNK1 à activer l’expression et la phosphorylation de NCC, même en absence de WNK4 [8]. Ces études in vivo sont donc en faveur d’une activation de NCC par L-WNK1 qui dépend de SPAK mais qui est indépendante de WNK4. La controverse WNK1 : une simple erreur technique ! Afin de comprendre les divergences observées entre les résultats obtenus in vitro et ceux observés in vivo, nous avons à nouveau examiné les effets de L-WNK1 en utilisant différents systèmes d’étude in vitro. Huit exons de WNK1 (exons 4a, 8b, HSN2, 9, 11, 12, 26a et 26b) sont soumis à un épissage alternatif [16]. Chez l’homme et la souris, le variant L-WNK1-∆11, qui ne contient pas l’exon 11, est le variant majoritaire dans le néphron (il représente environ 70 % de tous les variants de L-WNK1) [16]. La grande majorité des études concernant la régulation de NCC par L-WNK1 in vitro a paradoxalement été réalisée en utilisant le variant L-WNK1-∆11-12, qui ne représente en fait, dans le rein, que 20 % des transcrits L-WNK1. Nous avons donc étudié la régulation de NCC par l’ensemble des variants humains, en utilisant les cellules HEK293 (human embryonic kidney)5, et l’œuf de xénope [8]. De façon surprenante, tous les variants de L-WNK1 sont susceptibles, après transfection dans ces cellules, d’activer le cotransporteur NCC. Le variant conférant la plus grande activation est cependant L-WNK1-∆11. L’absence d’activation de NCC par L-WNK1 observée dans les études précédentes n’est donc pas le résultat de l’utilisation d’un « mauvais variant ». Il est important de noter que ces travaux ont été réalisés avec le même ADNc de L-WNK1-∆11-12 que celui qui a été sous-cloné lors de l’identification de WNK1, en 2000, à partir d’une banque d’ADNc de cerveau de rat [6]. Pour notre étude, nous avons sous-cloné les différents variants à partir de tissus humains. L’analyse par séquençage des ADNc d’homme et de rat correspondant au variant L-WNK1-∆11-12 a mis en évidence une mutation dans la partie C-terminale de la protéine du rat (p.Gly210Ser) par rapport à l’homme. Cette mutation est le résultat d’une erreur de lecture par la Taq polymérase6, réalisée lors du sous-clonage de L-WNK1 à partir de la banque d’ADNc. Elle ne correspond pas à une variation d’espèce puisque le séquençage du génome du rat met en évidence une glycine à cette position, comme dans toutes les autres espèces, et non une sérine. La mutation de la glycine en sérine altère l’activité du variant et empêche l’activation de NCC. Le remplacement du résidu sérine par une glycine permet de restaurer l’activation de NCC par L-WNK1-∆11-12 de rat et, à l’inverse, la mutation de l’ADNc du variant humain prévient l’activation de NCC [8].Ces résultats confirment donc les résultats obtenus avec les modèles murins. Ils valident que L-WNK1 est un activateur puissant de NCC in vitro et in vivo. Ils montrent également l’importance d’utiliser le variant approprié pour étudier la fonction d’un gène dont le messager peut subir plusieurs épissages alternatifs et donner ainsi naissance à plusieurs variants de la protéine. En effet, dans notre étude, le niveau d’activation de NCC dépend du variant utilisé, le niveau le plus élevé étant obtenu avec le variant le plus exprimé dans le rein. | ||||||
Les études menées dans l’œuf de xénope ou les cellules en culture ont montré que WNK4 est un inhibiteur de NCC et que cette inhibition dépend de son activité kinase [9, 10]. Pourtant, les souris porteuses d’une inactivation de WNK4 présentent une diminution de l’abondance et de la phosphorylation de NCC [17]. WNK4 serait donc un activateur de NCC in vivo. Une deuxième série d’expériences a remis en question les résultats obtenus in vitro. Les mutations identifiées dans le locus WNK4 chez les patients FHHt sont des mutations ponctuelles faux-sens7, [5]. Elles sont pratiquement toutes localisées dans un petit motif de la protéine d’une dizaine d’acides aminés, présent dans toutes les kinases de la famille WNK. Les conséquences fonctionnelles de ces mutations sont longtemps restées indéterminées. Ce motif est situé en aval d’un domaine coiled-coil, généralement décrit comme un motif responsable de l’oligomérisation des protéines (c’est-à-dire leur appariement) [18]. Ces mutations pourraient donc modifier l’interaction de WNK4 avec ses partenaires. Cette hypothèse a été confirmée grâce à l’identification de deux nouveaux gènes dont des mutations sont responsables de la FHHt [19, 20], KLHL3 (Kelch-like 3) et Cullin3 [35] (→). (→) Voir la Nouvelle de H. Louis-Dit-Picard et al., m/s n° 8-9, août-septembre 2012, page.703 Les protéines produites par ces gènes forment un complexe ubiquitine-ligase dans lequel KLHL3 recrute les substrats pour qu’ils soient ubiquitinés, entraînant ainsi leur dégradation protéosomale8. WNK4 est l’un de ces substrats [21–23]. Les mutations touchant WNK4, responsables de la FHHt, empêchent son interaction avec KLHL3, ce qui conduit à une augmentation de l’abondance de la protéine WNK4. Ces résultats ont été confirmés in vivo. L’expression de WNK4 est en effet augmentée dans le rein de souris porteuses d’une des mutations de WNK4 associées à la FHHt, et qui présentent toutes les signes de la FHHt [22]. La protéine WNK4 mutée aurait donc la même activité que la protéine sauvage. Cette hypothèse a été vérifiée in vivo. Des souris surexprimant la forme sauvage de WNK4 présentent en effet une augmentation de l’expression et de la phosphorylation de NCC ainsi qu’une FHHt [22]. L’ensemble de ces études permet donc de conclure que WNK4 est un activateur fort et essentiel de NCC in vivo. Comment expliquer alors que cette activation n’ait pas été observée in vitro ? L’explication la plus simple est celle d’une différence existant entre les systèmes utilisés pour les expériences in vitro (cellules HEK et œuf de xénope) et les cellules du DCT in situ, qui influencerait l’activité de WNK4. Plusieurs études avaient suggéré que l’activité des WNK était modulée par des changements de concentration ionique, notamment en chlorure et en potassium. Plus récemment, Piala et al. ont montré que L-WNK1 est une kinase dont l’activité est sensible au chlorure [24]. Elle est en effet plus active lorsque la concentration en chlorure est faible. L’activation de L-WNK1 requiert la phosphorylation d’une sérine (S382) située dans la boucle d’activation du domaine kinase (activation loop) [25], qui est plus importante lorsque la concentration en chlorure est faible [24]. Des études de cristallographie ont mis en évidence que le chlorure se fixe au domaine kinase, maintenant la protéine dans une conformation inactive. Un résidu leucine (L379) joue un rôle crucial dans la formation du site de fixation pour le chlorure dans le domaine kinase. En effet, sa mutation en phénylalanine diminue la sensibilité de L-WNK1 au chlorure [24]. La concentration en chlorure retrouvée dans les modèles cellulaires in vitro est plus élevée que celle observée dans les cellules du DCT (40-50 mM [7] versus 10-20 mM [26]). Les effets divergents de WNK4 sur NCC pourraient donc résulter de cette différence de concentration intracellulaire en chlorure ([Cl-]i) entre les deux systèmes cellulaires. Nous avons testé l’activité de NCC dans les ovocytes de xénope incubés dans un milieu standard ([Cl-]i ≈ 50 mM) ou dans un milieu hypotonique dépourvu de chlorure ([Cl-]i ≈ 35 mM). La co-injection de NCC et de WNK4 dans l’œuf de xénope en conditions standard résulte en une diminution de l’absorption de sodium par NCC. Cependant, lorsque la concentration intracellulaire de chlorure est abaissée expérimentalement, WNK4 active NCC (Figure 2). L’activation de WNK4, comme pour L-WNK1, dépend de la phosphorylation de sa boucle d’activation, qui augmente lorsque la concentration intracellulaire de chlorure diminue (Figure 2). La mutation de la leucine présente dans le site de fixation du chlorure permet à WNK4 d’activer NCC dans les conditions standard en absence de déplétion en chlorure. WNK4 est donc une kinase sensible au chlorure. Ces observations expliquent donc vraisemblablement la dualité des effets activateurs et inhibiteurs que WNK4 peut avoir sur NCC en fonction de la concentration intracellulaire de chlorure.
Le fait que L-WNK1 puisse activer NCC dans l’œuf de xénope en conditions standard alors qu’il est nécessaire d’abaisser la concentration intracellulaire de chlorure ([Cl-]i) pour obtenir le même effet avec WNK4 suggère que cette dernière est plus sensible à la concentration de chlorure que L-WNK1. Cette hypothèse a été très récemment confirmée par une nouvelle étude. Terker et al ont en effet montré que l’activité kinase de WNK4 diminue progressivement lorsque la concentration intracellulaire en chlorure augmente de 10 à 40 mM, alors que celle de L-WNK1 est stable aux mêmes concentrations et ne commence à diminuer que lorsque cette concentration atteint 60 mM [27]. | ||||||
L’ensemble des données récentes que nous avons présentées a permis d’établir un nouveau modèle de régulation de NCC par WNK1 et WNK4. Elles réconcilient les observations in vivo et in vitro (Figure 3). Il reste désormais à déterminer les conditions physiologiques dans lesquelles cette voie de régulation intervient. Deux études suggèrent qu’elle pourrait être mise en jeu pour adapter l’excrétion de potassium en réponse à des modifications de la quantité de cet élément dans l’alimentation.
Plusieurs études réalisées chez le rat et la souris ont ainsi montré que l’expression de NCC varie en fonction de la teneur en potassium des aliments : elle est augmentée lorsque le régime est pauvre en potassium et diminuée lorsque le régime est riche en potassium [28] (Figure 3). La diminution de l’expression de NCC dans cette dernière condition augmente la quantité de sodium délivrée au tubule connecteur (CNT) et au canal collecteur cortical (CCD), situés en aval du DCT. Ceci stimule l’activité du canal épithélial sodique (ENaC), exprimé dans ces segments du néphron (Figure 1). La réabsorption de sodium par ENaC est électrogénique. Elle est accompagnée d’une sécrétion de potassium par le canal potassique ROMK (renal outer medullary potassium channel), ce qui permet l’excrétion du potassium en excès (Figure 1). À l’inverse, lors d’une restriction en potassium, la majorité du sodium est réabsorbée par NCC dans le DCT, ce qui diminue la réabsorption de sodium par ENaC et la sécrétion de potassium par ROMK. Ce mécanisme pourrait expliquer l’hyperkaliémie observée chez les patients FHHt. La question restait de comprendre comment est contrôlé le niveau d’expression de NCC dans ces deux conditions. Terker et al. ont récemment démontré que la concentration extracellulaire de potassium ([K+]e) a une influence directe sur la concentration intracellulaire de chlorure ([Cl-]i) [29]. Ainsi, une [K+]e basse provoque une diminution de la [Cl-]i et stimule l’expression de NCC via la voie de signalisation WNK1-SPAK (Figure 3). La modification de la [Cl-]i par la [K+]e fait intervenir le canal potassique Kir4.1, également appelé KCNJ10 (potassium channel, inwardly rectifying subfamily J, member 10). Ce canal est exprimé au pôle basolatéral des cellules du DCT, du CNT et du CCD. En sécrétant du potassium, ce canal assure le recyclage de cet ion pour la pompe Na+/K+-ATPase, qui permet la sortie du sodium réabsorbé par NCC. Cette sécrétion de potassium permet également d’établir un gradient électrochimique propice qui favorise l’efflux des ions chlorure réabsorbés par NCC via le canal chlorure CLCKb (renal chloride channel b)/Barttine, en maintenant cet ion hors de l’équilibre (Figure 4). La conductance potassique dans le tube contourné distal dépendant du seul canal Kir4.1, une modulation de l’activité de ce canal est susceptible d’influencer largement l’efflux de chlorure en faisant varier la différence de potentiel membranaire. Cette relation entre activité de Kir4.1 et efflux des ions chlorure a été récemment démontrée par l’équipe de W.H. Wang, grâce à la caractérisation d’un modèle d’inactivation génique [30]. Des expériences de patch-clamp 9, ont en effet montré que la conductance au chlorure baso-latérale de DCT isolés de souris Kir4.1-/- est inférieure à celle des DCT de souris sauvages. Cette diminution de conductance devrait conduire à une augmentation de la concentration en chlorure intracellulaire. Si cela n’a pas été démontré chez la souris, Terker et al. ont montré que c’est le cas dans des cellules HEK293 exprimant un mutant nul10, du gène Kir4.1 [29]. En conséquence, l’augmentation de la [Cl-]i devrait à son tour inhiber l’activation de SPAK par les WNK et donc l’expression et la phosphorylation de NCC (Figure 4). Cette hypothèse a été vérifiée chez la souris Kir4.1 -/-. Ces animaux présentent en effet une diminution de l’expression de SPAK et de NCC [30]. Ces résultats permettent d’expliquer comment des mutations inactivatrices du gène codant Kir4.1 peuvent être à l’origine du syndrome EAST (epilepsy, ataxia, sensorineural deafness, and tubulopathy)/SeSAME (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance), un syndrome qui touche de nombreux organes et dont les manifestations rénales sont semblables à celles observées chez les patients atteints du syndrome de Gitelman causé, lui, par des mutations inactivatrices de NCC [31, 32].
| ||||||
L’ensemble de ces études a donc permis de réconcilier les données in vivo et in vitro, obtenues ces dix dernières années, sur la régulation de l’activité de NCC par les WNK. Elles ont surtout permis de commencer à comprendre comment la voie WNK-SPAK-NCC intervient en conditions physiologiques, notamment lors de variations de l’apport en potassium. Il reste cependant encore de nombreux points à éclaircir pour comprendre pleinement le rôle complexe joué par ces kinases dans la régulation coordonnée de la balance du sodium et du potassium. Une question cruciale est notamment la mesure de la concentration intracellulaire de chlorure et de ses variations dans ces différentes conditions. | ||||||
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. | ||||||
3
Les thiazidiques ont été développés initialement pour leur propriété d’inhibition de l’anhydrase carbonique mais leur effet diurétique s’est révélé supérieur. Ils sont constitués par la fusion entre un noyau de benzène et un noyau de thiadiazine (comportant 2 atomes d’azote et 1 atome de soufre).
4
Le xénope est un amphibien, eucaryote. Il présente l’avantage de pondre beaucoup d’œufs, autant d’embryons larges et facilement manipulables. Ses embryons représentent un bon modèle d’étude du transport ionique, après surexpression des protéines d’intérêt.
5
La lignée de cellules rénales embryonnaires humaines HEK293 est très utilisée pour les expériences de transfection de gène afin d’étudier la fonctionnalité de la protéine correspondante.
6
La Taq polymérase est une ADN polymérase utilisée pour la duplication de l’ADN au cours de la réaction de PCR (polymerase chain reaction).
7
La mutation faux-sens occasionne un changement au niveau d’un nucléotide d’un codon. Elle provoque une modification de l’acide aminé qui lui est associé. L’activité de la protéine traduite peut en être modifiée.
8
Le système ubiquitine/protéasome joue un rôle majeur dans la protéolyse intracellulaire. De nombreux composants de ce système sont des complexes protéiques présentant dans une même structure, le protéasome, plusieurs activités enzymatiques, les enzymes d’ubiquitinylation.
| ||||||
1.
Berghoff RS, Geraci AS. The influence of sodium chloride on blood pressure . Br Med J. 1929; ; 56 : :395.–397. 2.
Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. DASH-sodium collaborative research group . N Engl J Med. 2001; ; 344 : :3.–10. 3.
Guyton AC, Coleman TG, Cowley AV, Jr, et al. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension . Am J Med. 1972; ; 52 : :584.–594. 4.
Gordon RD, Klemm, SA, Tunny TJ et al. Gordon’s syndrome: a sodium-volume-dependent form of hypertension with a genetic basis . In: Brenner JHLaBM, ed., Hypertension: patholgy, diagnosis and management . New York: : Raven Press; , 1995 : :2111.–2113. 5.
Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases . Science. 2001; ; 293 : :1107.–1112. 6.
Xu B, English JM, Wilsbacher JL, et al. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II . J Biol Chem. 2000; ; 275 : :16795.–16801. 7.
Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, et al. The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride . J Am Soc Nephrol. 2014; ; 26 : :1781.–1786. 8.
Chavez-Canales M, Zhang C, Soukaseum C, et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4 . Hypertension. 2014; ; 64 : :1047.–1053. 9.
Wilson FH, Kahle KT, Sabath E, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4 . Proc Natl Acad Sci USA. 2003; ; 100 : :680.–684. 10.
Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport . J Clin Invest. 2003; ; 111 : :1039.–1045. 11.
Delaloy C, Lu J, Houot AM, et al. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform . Mol Cell Biol. 2003; ; 23 : :9208.–9221. 12.
Vidal-Petiot E, Elvira-Matelot E, Mutig K, et al. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron . Proc Natl Acad Sci USA. 2013; ; 110 : :14366.–14371. 13.
Golbang AP, Cope G, Hamad A, et al. Regulation of the expression of the Na/Cl cotransporter (NCCT) by WNK4 and WNK1: evidence that accelerated dynamin-dependent endocytosis is not involved . Am J Physiol Renal Physiol. 2006; ; 291 : :F1369.–F1376. 14.
Moriguchi T, Urushiyama S, Hisamoto N, et al. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1 . J Biol Chem. 2005; ; 280 : :42685.–42693. 15.
Zambrowicz BP, Abuin A, Ramirez-Solis R, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention . Proc Natl Acad Sci USA. 2003; ; 100 : :14109.–14114. 16.
Vidal-Petiot E, Cheval L, Faugeroux J, et al. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms . PLoS One. 2012; ; 7 : :e37751.. 17.
Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, et al. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process . Proc Natl Acad Sci USA. 2012; ; 109 : :7929.–7934. 18.
Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif . Trends Cell Biol. 2001; ; 11 : :82.–88. 19.
Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities . Nature. 2012; ; 482 : :98.–102. 20.
Louis-Dit-Picard H, Barc J, Trujillano D, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron . Nat Genet. 2012; ; 44 ((456–60)) : :S1.–S3. 21.
Shibata S, Zhang J, Puthumana J, et al. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4 . Proc Natl Acad Sci USA. 2013; ; 110 : :7838.–7843. 22.
Wakabayashi M, Mori T, Isobe K, et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension . Cell Rep. 2013; ; 3 : :858.–868. 23.
Wu G, Peng JB. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation . FEBS Lett. 2013; ; 587 : :2099.–2104. 24.
Piala AT, Moon TM, Akella R, et al. Chloride sensing by WNK1 involves inhibition of autophosphorylation . Sci Signal. 2014; ; 7 : :ra41.. 25.
Zagorska A, Pozo-Guisado E, Boudeau J, et al. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress . J Cell Biol. 2007; ; 176 : :89.–100. 26.
Boettger T, Hubner CA, Maier H, et al. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4 . Nature. 2002; ; 416 : :874.–878. 27.
Terker AS, Zhang C, Erspamer KJ, et al. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis . Kidney Int. 2015 ; doi : 10.1038/ki.2015.289. 28.
Vallon V, Schroth J, Lang F, et al. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1 . Am J Physiol Renal Physiol. 2009; ; 297 : :F704.–F712. 29.
Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride . Cell Metab. 2015; ; 21 : :39.–50. 30.
Zhang C, Wang L, Zhang J, et al. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) . Proc Natl Acad Sci USA. 2014; ; 111 : :11864.–11869. 31.
Reichold M, Zdebik AA, Lieberer E, et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function . Proc Natl Acad Sci USA. 2010; ; 107 : :14490.–14495. 32.
Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10 . Proc Natl Acad Sci USA. 2009; ; 106 : :5842.–5847. 33.
Eladari D, Chambrey R, Leviel F. Identification d’une nouvelle cible des diurétiques thiazidiques dans le rein . Med Sci (Paris). 2010; ; 26 : :549.–552. | ||||||




