
Vignette (© AFM - Téléthon).
| |||
Med Sci (Paris). 33: 57. doi: 10.1051/medsci/201733s112.Génétique : Intérêt du NGS dans un cas atypique de LGMD liée à l’alphadystroglycane 1Centre de Recherche en Myologie, Sorbonne Universités, UPMC – Inserm UMRS 974, Institut de Myologie, Groupe Hospitalier Pitié-Salpêtrière, Paris, France Corresponding author. | ||
Vignette (© AFM - Téléthon). | ||
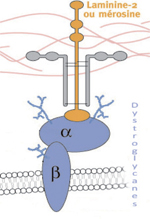
Des défauts de l’O-glycosylation de l’apha-dystroglycane (α-DG) sont responsables d’un large éventail de maladies neuromusculaires allant des dystrophies musculaires congénitales (DMC) sévères associées à un développement anormal du cerveau et des yeux, aux formes légères de dystrophie des ceintures. L’étude présentée ici [1] rapporte le cas d’une patiente ayant développé un déficit et une fonte musculaire de la ceinture pelvienne devenues symptomatique à l’âge de 42 ans. Le séquençage de l’exome (régions codantes de tous les gènes) a révélé la présence d’une substitution à l’état homozygote (c.131T > G (p.Leu44Pro)) dans le gène DPM3. Ce dernier code la sous-unité 3 de la dolichol-P-mannose (DPM) synthase, entraînant une réduction de moitié de l’activité enzymatique. La diminution de DPM disponible comme substrat essentiel donneur pour les O-mannosyltransférases (POMT) 1 et 2 explique le défaut de la O-glycosylation de l’α-DG dans le muscle squelettique. Les résultats de cette étude démontrent que des mutations du gène DPM3 peuvent être à l’origine induire d’une présentation clinique de dystrophie des ceintures peu grave et isolée, sans cardiomyopathie associée. Commentaire Les dystroglycanes (DG), alpha et bêta, sont les éléments centraux du complexe des glycoprotéines associées à la dystrophine et permettent notamment d’assurer le lien entre le cytosquelette intracellulaire et la matrice extracellulaire. La sous-unité α-DG est très fortement glycosylée et pas moins de 18 gènes sont impliqués dans les α-DGpathies dont le point commun est un défaut de la glycosylation de cette sous-unité [2].Dans l’étude présentée ici, la présentation clinique et les résultats d’immunomarquages sur la biopsie musculaire avec l’anticorps le plus communément utilisé pour détecter l’α-DG (IIH6), avaient d’emblée orienté vers un diagnostic de LGMD lié à l’α-DG. Le séquençage de l’exome de la patiente, dans le cadre du projet MYO-SEQ [3] a permis d’identifier un variant homozygote dans legène DPM3 codant une enzyme intermédiaire dans la chaîne de O-glycosylation. Toutes les prédictions (fréquence, conséquences au niveau protéique) suggéraient fortement la pathogénicité du variant, qui de plus co-ségrégeait parfaitement avec la maladie dans la famille. D’un point de vue fonctionnel, la synthèse de dolichol-P-mannose (DPM) est diminuée de moitié dans les fibroblastes de la patiente. Le DPM étant un substrat important requis dans la synthèse des chaînes N- ou O- glycanes, le déficit de glycosylation de l’α-DG détecté par l’anticorps IIH6 se trouve donc explicité. Des mutations des trois sous-unités de la DPM synthase avaient précédemment été rapportées, associant des présentations cliniques différentes de celle décrite ici, de moindre sévérité. Ce travail montre à nouveau l’intérêt du NGS pour élucider des cas atypiques ou sans solution évidente. | ||
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. | ||
1.
Lefeber DJ, Xu L, Lek M, et al. A homozygous DPM3 mutation in a patient with alpha-dystroglycan-related limb girdle muscular dystrophy . Neuromuscular Disord. 2017; Jul 17. pii: S0960–8966(17)30436–4. | ||
