 |
| |||
Med Sci (Paris). 33: 58–60. doi: 10.1051/medsci/201733s113.Préclinique | ||
| ||
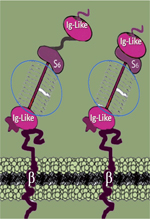
Structural flexibility of the human α-dystroglycan Dominique Mornet Résumé En 1992, il a été établi qu’un seul ARN messager, issu du gène DAG1, produisait une protéine unique qui allait être maturée pour donner finalement 2 glycoprotéines dénommées DAG-1 et DAG-2. Ces dernières seront par la suite désignées comme les protéines 156-DAG et 43-DAG puis cataloguées comme étant respectivement les formes α et β- (DG). L’entité β-DG est ancrée dans la membrane cellulaire et accroche d’une part l’α-DG qui se situe dans la matrice extracellulaire et d’autre part la dystrophine située dans le cytoplasme de la cellule.L’étude menée dans ce travail [1] clone, exprime et purifie un segment N-terminal (résidus 52-315) correspondant à la séquence humaine de l’α-DG. Une telle protéine recombinante est alors soumise à diverses approches expérimentales : (1) des expériences de fluorimétrie à balayage différentiel (excitation 470-505 nm, et émission 540-700 nm) qui sont répétées 3 fois en utilisant un gradient de température de 20 à 90°C ; (2) des analyses de protéolyses limitées qui utilisent une batterie de protéases de spécificités différentes ; (3) une mise au point de conditions originales de cristallisation pour de nouvelles données via la technique de diffusion des rayons X aux petits angles. La structure cristallographique de l’α-DG a ainsi été obtenue avec une résolution de 1,8 Angströms. En appliquant ces différentes méthodes, une relative flexibilité dans la structure de la version humaine de l’α-DG est mise en évidence. Ce lien flexible dont la conformation est l’objet de l’étude présentée, est visualisé avec deux conformations différentes permettant une connexion entre la structure de type Immunoglobuline (Ig = résidus 62-160) et le domaine qui ressemble à une petite sous-unité ribosomale dite « S6 » (correspondant aux résidus 182-305). L’ensemble des résultats conforte des études similaires, déjà menées avec la séquence de l’α-DG exprimée chez la souris, grâce auxquelles les auteurs avaient préalablement acquis leur expertise. Cette nouvelle étude démontre ainsi la plasticité moléculaire d’une partie N-terminale de la version humaine de l’α-DG. Cette plasticité pourrait avoir un rôle important pour les interactions fonctionnelles impliquant une liaison avec ses nombreux autres partenaires au niveau de la matrice extracellulaire. Commentaire Au fur et à mesure des découvertes sur la structure/la biologie des dystroglycanes, il a été montré qu’un peptide unique de 98 kDa est obtenu à partir de l’ARN messager de 5,8 kb. Un clivage enzymatique spécifique donne alors deux chaînes peptidiques de 72 et 26 kDa. Une maturation impliquant plusieurs types de glycosylations va générer les entités de 156 et 43 kDa. Ces glycosylations post-traductionnelles sont relativement importantes et la nature des groupements glycosylés va dépendre de la spécificité tissulaire. Progressivement, le portrait-robot des dystroglycanes est revisité avec d’une part l’identification précise du site de maturation alpha-bêta (séquence GLY-653-SER-654), et d’autre part les aspects mécanistiques de la conformation finale de la forme α-DG résultant des divers types de sucres associés à la glycosylation extensive de cette protéine ainsi que les défauts de glycosylation associés. C’est notamment le cas de la mutation p.T192M sur la forme humaine de l’α-DG pour son impact spécifique dans l’inhibition de la glycosylation par la « Glycosyltransferase-like protein LARGE1 » dite LARGE [2]. Depuis 2004, les données cristallographiques de la partie N-terminale de α-DG sont connues et une analyse précise de la zone comprenant les résidus 30 à 315 indiquait clairement la présence des 2 sous-domaines Ig (aa 62–160) et S6 (aa 182–305). De plus, une étude originale de 2015 rapporte une tendance à une protéolyse partielle en présence de la métalloprotéinase MMP2 de cette zone [3]. Il est également bien établi que l’α-DG possède une grande affinité pour les laminines et autres protéines contenant un domaine LG (laminine globulaire) et diverses protéoglycanes. Il représente ainsi un module autonome qui peut être libéré par la protéolyse dirigée par les furines dans l’espace extracellulaire et/ou les fluides corporels. Le domaine N-terminal Ig a été proposé comme ayant des fonctions supplémentaires dans le processus de la glycosylation impliqué dans la voie de maturation du peptide précurseur des dystroglycanes comme le souligne une récente revue sur le sujet [4]. On peut y ajouter une potentielle fonction dans la signalisation trans-synaptique spécifique d’un sous-type inter-neuronal, révélant une corrélation entre la diversité moléculaire pré-synaptique et post-synaptique. Pour toutes ces raisons, il paraît important de bien établir le degré de flexibilité qui existe dans cette portion N-terminale de l’a-DG pour mieux en comprendre la structure nécessaire à une fonctionnalité totale. | ||
Duchenne muscular dystrophy: efficacy of systemic AAV-mediated gene transfer in a dog model Jean-Thomas Vilquin Résumé L’injection chez le chien GRMD, un des modèles animaux de la myopathie de Duchenne, d’un vecteur viral contenant un gène codant une microdystrophine stabilise ou améliore durablement l’état clinique des animaux. L’injection est réalisée par voie locale ou systémique, sans immunosuppression, et les effets, mesurés par de nombreuses techniques complémentaires, persistent jusqu’à deux ans au moins [1].La myopathie de Duchenne est provoquée par des mutations du gène de la dystrophine causant l’absence de son expression et/ou de sa fonctionnalité. Un microgène canin optimisé, appelé microdystrophine (cMD1), a été développé pour permettre l’empaquetage par un vecteur adéno-associé (rAAV2/8), particulièrement efficace pour le ciblage musculaire squelettique et cardiaque. Deux voies d’injections ont été testées successivement chez le chien GRMD, en absence de toute immunosuppression. La voie locorégionale par perfusion de membre sous légère surpression, et la voie systémique intraveineuse ont toutes deux permis une distribution efficace des vecteurs. Les injections ont été bien tolérées à toutes doses (1 x 1013, 2 x 1013 et 1014 génomes viraux par kg), n’entraînant pas de toxicité hématologique, biologique, rénale, hépatique. L’expression de dystrophine tronquée a été mise en évidence par des techniques complémentaires (immunohistochimie, Western-blot, Q-PCR). Ces traitements ont permis la réduction de la régénération musculaire et des dépôts de collagène (fibrose), une réduction oedémateuse, de l’inflammation et de la nécrose, une stabilisation ou une amélioration de la force musculaire, l’ensemble suggérant une réduction de la pathologie dystrophique chez les chiens. Des variations ont été rapportées en fonction des voies, des doses, et des territoires analysés. Par voie locorégionale, les effets sont particulièrement marqués au territoire d’injection, mais sont également observés dans des zones en aval. L’administration systémique, plus globale, améliore à long terme le phénotype clinique : les chiens restent ambulants, ont un meilleur score clinique, et survivent plus longtemps par rapport aux animaux non traités. De manière très intéressante, une réponse immunitaire humorale est générée contre le produit de transgène, mais de manière transitoire et non délétère, et disparaît au cours du temps. Une réponse humorale forte (anticorps neutralisants) est développée contre les capsides des vecteurs. Aucune réponse à médiation cellulaire n’a été observée. Ces résultats vont dans le sens d’une stabilisation ou d’une amélioration dose-dépendante du statut clinique et ouvrent la voie à des essais cliniques chez l’Homme. Commentaire Ces travaux représentent une synthèse, un aboutissement d’années de recherche consacrées à comprendre puis à circonvenir les nombreuses complexités liées à la myopathie de Duchenne et à son approche thérapeutique. La conception d’un transgène canin « microdystrophine » optimisé et finement régulé est indissociable des études structurales et fonctionnelles de cette protéine et de son gène gigantesques, et de la biologie moléculaire des contrôles d’expression. Mais la définition d’un petit transgène résulte aussi de la définition rigoureuse des meilleurs vecteurs et de leur biologie. L’efficacité du concept de supplémentation génique a dû être prouvée successivement in vitro, puis chez le petit animal par différentes voies, puis finalement chez le gros animal, qui reproduit plus fidèlement la physiopathologie et la sensibilité immunologique humaines, ce qui nécessite une connaissance approfondie des subtilités de ces modèles. L’efficacité des vecteurs est particulièrement remarquable, et l’absence de réaction immunologique peut lever des obstacles thérapeutiques, éthiques, et battre en brèche certains dogmes. Plusieurs publications confirment et renforcent l’intérêt thérapeutique de ces approches d’injection systémique de vecteurs AAV. Ainsi, l’expression de dystrophine peut être promue par la technique du saut d’exon en utilisant un transgène spécifique véhiculé par voie locorégionale au sein d’un vecteur AAV chez le chien GRMD [1]. De même, en utilisant un transgène codant cette fois-ci la myotubularine, il est possible de corriger la totalité de la musculature et d’améliorer durablement la survie, les scores neurologiques, la force, la respiration dans un modèle canin déjà symptomatique de myopathie myotubulaire liée au chromosome X (XLMT1), sans déclencher de réaction immunologique délétère [2]. Des questions peuvent certes encore se poser : l’effet s’atténue-t-il au très long cours, notamment du fait de la croissance corporelle, de la dilution des génomes viraux, de l’inactivation des promoteurs ? La réponse immunologique anti-virale persiste-t-elle, décline-t-elle, peut-elle être contrôlée ? Est-il possible d’envisager une ré-administration, ou le traitement de sujets sensibilisés aux AAV ? Il est cependant permis d’espérer qu’une étape importante est franchie vers la définition de nouvelles stratégies d’essais cliniques. | ||
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. | ||
1.
Covaceuszach S, Bozzi M, Bigotti MG, et al. Structural flexibility of human α-dystroglycan . FEBS Open Bio. 2017; ; 7 : :1064.–1077. 2.
Kanagawa M, Saito F, Kunz S, et al. Molecular recognition by LARGE is essential for expression of functional dystroglycan . Cell. 2004; ; 117 : :953.–964. 3.
Sbardella D, Sciandra F, Gioia M, et al. α-dystroglycan is a potential target of matrix metalloproteinase MMP-2 . Matrix Biol. 2015; ; 41 : :2.–7. 4.
Brancaccio A, Adams JC. An evaluation of the evolution of the gene structure of dystroglycan . BMC Res Notes. 2017; ; 10 : :19.. | ||
