



Le syndrome de Schwartz-Jampel
Résumé
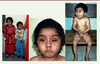
Le syndrome de Schwartz-Jampel (SJS, OMIM #255800) est une affection génétique ultra-rare définie par des manifestations myotoniques et des anomalies ostéo-articulaires. Transmis selon un mode autosomique récessif, sa prévalence est plus élevée dans les zones de forte endogamie. La découverte du gène
HSPG2
codant une protéine de la lame basale a permis de mieux en délimiter les contours nosologiques. Le diagnostic est généralement très fortement suspecté cliniquement puis confirmé en biologie moléculaire. Le traitement reste à ce jour essentiellement symptomatique. The Schwartz-Jampel syndrome (SJS, OMIM #255800) is an ultra-rare genetic disease characterized by myotonic manifestations combined with bone and cartilage abnormalities. Following an autosomal recessive mode of inheritance, its prevalence is more significant in highly-inbred areas. The unraveling of the
HSPG2
gene encoding a protein of the basal lamina enabled a better nosological delineation of the syndrome. The diagnosis is usually strongly suspected at the clinical level and then confirmed by molecular biology. To date, the treatment remains essentially symptomatic.
Pour citer ce document
Urtizberea, J. Andoni ; Severa, Gianmarco ; Ropars, Juliette ; Malfatti, Edoardo ; Le syndrome de Schwartz-Jampel, Med Sci (Paris), Vol. 39, N° HS ; p. 37-46 ; DOI : 10.1051/medsci/2023133