



La polykystose autosomique dominante : progrès cliniques et génétiques
Résumé
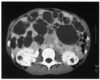
La polykystose autosomique dominante est la plus fréquente
des maladies génétiques rénales. Elle est caractérisée
par le développement de multiples kystes dans le rein,
lié à une sécrétion liquidienne dans des structures composées
de cellules relativement immatures. La fibrose interstitielle
et des phénomènes apoptotiques contribuent probablement
à l'altération de la fonction rénale. Celle-ci est
déterminée par des facteurs génétiques dont au moins
deux gènes, PKDJ et PKD2, mais aussi le sexe et peut-être
la régulation de la pression artérielle. Les manifestations
extrarénales de la maladie comportent fréquemment une
polykystose hépatique, habituellement sans gravité intrinsèque,
et des anomalies vasculaires cérébrales à type d'anévrismes.
Le gène responsable de 85 % des cas, PKDJ, code
pour une protéine intégrale de la membrane plasmique
dont le domaine extracellulaire pourrait être impliqué dans
des interactions avec d'autres cellules ou avec la matrice
extracellulaire. La protéine PKD2, elle aussi transmembranaire,
semble être un canal ionique ou un pore dont la
fonction pourrait être réglée par PKD 1. Autosomal dominant polycystic kidney disease (ADPKD), the most frequent inherited kidney disease, usually manifest in adulthood, is characterized by the development of multiple renal cysts variably associated with extra-renal abnormalities. Pathophysiologic studies have shown that expansion of kidney cysts results from transepithelial fluid secretion and cellular proliferation of relatively immature cells, in association with remodeling of the tubular basement membrane. Interstitial fibrosis and widespread apoptosis likely contribute to the loss of renal function. Clinical determinants of progression to renal insufficiency are the genetic form of the disease, gender and probably blood pressure level. Liver cysts, occurring in 80% of patients by age 50, are usually asymptomatic. Some patients with massive polycystic liver may require cyst fenestration and/or resection. Cerebral aneurysm is detected in 8% of patients overall, and in 16% of those with a family history of cerebral aneurysm. It may rupture and lead to subarachnoid haemorrhage. Screening for cerebral aneurysm is recommended in young patients with a positive family history. The gene responsible for 85% of the cases (PKD1), has been identified in chromosome region 16p13. It covers 52 kb and includes 46 exons giving a transcript of 14.5 kb coding for a protein of 4,203 amino acids, now called polycystin. Polycystin is probably an integral membrane protein with multiple extra-cellular domains that are involved in cell-cell and/or cell-matrix interactions. The gene accounting for the vast majority of the remaining cases (PKD2) has been identified in chromosome region 4q21. It codes for a protein of 968 amino acids, which appears to be a transmembrane protein, putatively functioning as an ion channel or pore. PKD1 could act as the regulator of PKD2 activity. Disruption of communication between matrix and cell by the mutated protein could account for the whole clinico-pathological features of ADPKD. Further understanding of both the function of PKD1 and PKD2 proteins and the cystic pathway should pave the way for therapeutic intervention. [References: 51]
Pour citer ce document
Pirson, Y ; Chauveau, D ; Watson, ML ; Zeier, M ; Breuning, M, La polykystose autosomique dominante : progrès cliniques et génétiques, Med Sci (Paris), 1997, Vol. 13, N° 1; p.37-44