| dc.contributor.author | Médigue, Claudine | - |
| dc.contributor.author | Bocs, Stéphanie | - |
| dc.contributor.author | Labarre, Laurent | - |
| dc.contributor.author | Mathé, Catherine | - |
| dc.contributor.author | Vallenet, David | - |
| dc.date.accessioned | 2014-02-14T12:31:11Z | |
| dc.date.available | 2014-02-14T12:31:11Z | |
| dc.date.issued | 2002 | fr_FR |
| dc.identifier.citation | Médigue, Claudine ; Bocs, Stéphanie ; Labarre, Laurent ; Mathé, Catherine ; Vallenet, David ; L’annotation in silico des séquences génomiques, Med Sci (Paris), 2002, Vol. 18, N° 2; p. 237-250 ; DOI : 10.1051/medsci/2002182237 | fr_FR |
| dc.identifier.issn | 1958-5381 | fr_FR |
| dc.identifier.uri | http://hdl.handle.net/10608/4906 | |
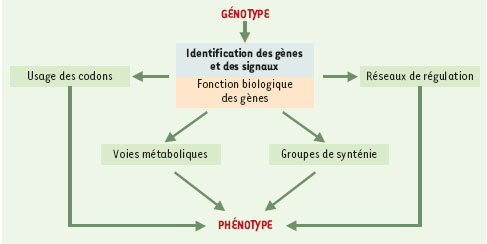
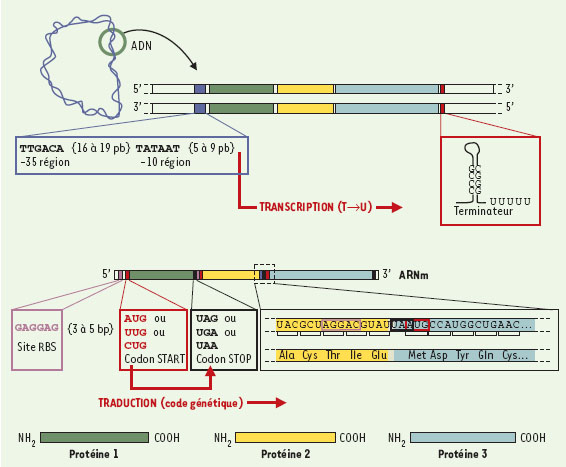
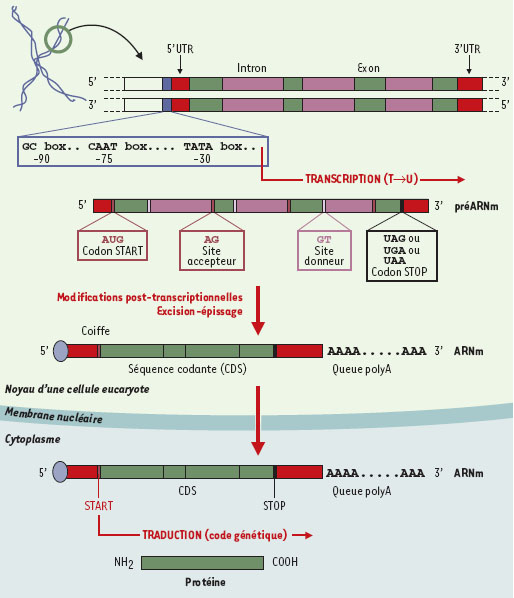
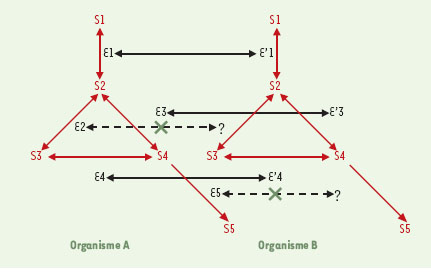
| dc.description.abstract | Depuis 1995, nous avons accès à l’information génétique complète d’un nombre croissant d’organismes vivants très divers. Cette explosion d’informations impose des changements profonds dans de nombreuses disciplines scientifiques, particulièrement en bio-informatique et en génétique moléculaire. L’un des plus importants défis est de prédire et d’annoter les fonctions de la plupart des produits de gènes de façon à la fois rapide et exhaustive, en tenant compte des interactions moléculaires entre les différents éléments prédits (expression de la régulation des gènes et données métaboliques). Au-delà de l’information fournie par la séquence complète des génomes, ces dernières analyses requièrent des données complémentaires issues de l’étude du transcriptome et du protéome. Aussi, de nouvelles infrastructures informatiques, intégrant différents niveaux d’annotation de séquences et de prédiction des fonctions biologiques, vont devenir indispensables. Cette revue est destinée à décrire les démarches permettant l’annotation in silico des séquences génomiques d’organismes procaryotes et euca-ryotes. Un regard spécifique est porté sur les problèmes auxquels se heurte tout annotateur, ainsi que les voies de recherches actuelles dans ce domaine. | fr |
| dc.description.abstract | For the first time in history, we have access to the entire genetic content of a growing number and variety of living organisms. This explosive growth of information is forcing changes in many scientific disciplines, particularly in computational biology and molecular genetics. One of the challenges is to predict and annotate the functions of the gene products as rapidly and completely as possible, taking into account both molecular interactions and higher cellular order processes. The first level of sequence annotation consists in gene finding and functional prediction of their products using similarities searching in protein databanks. This step remains easier in the context of procaryotic genome analysis, the gene structure of these organisms being much more simple than the one of eucaryotes. Predicting function from sequence using computational tools is generally done for each gene individually. Others levels of annotation, such as the identification of interactions between genomic elements characterized in the first step, are more difficult to achieve. If we currently best described the protein function in the context of molecular interactions, it will be possible in the near future to predict function in the context of higher order processes such as the regulation of gene expression, metabolic pathways and signaling cascades. Besides the information from the completely sequenced genomes, the latter analysis also uses additional information from proteomics and expression data. New infrastructures that integrate various levels of sequence annotation and function prediction are clearly required. This paper focuses on the various facets of the in silico sequence annotation, which is far from being perfect despite the fact that sequencing itself is highly automated and accurate, and despite the fact that (or maybe because…) sequence information is described in simple linear form, using a four-letter alphabet. There remains a long way to go until we are able to describe molecular processes quantitatively. However, there is no doubt that in silico sequence analysis is extremely powerful, and the generation of hypothesis derived by computational methods will be more and more often the first successful step in the design of in vivo/in vitro experiments. | en |
| dc.language.iso | fr | fr_FR |
| dc.publisher | EDK | fr_FR |
| dc.relation.ispartof | Repères : Lexique | fr_FR |
| dc.rights | Article en libre accès | fr |
| dc.rights | Médecine/Sciences - Inserm - SRMS | fr |
| dc.source | M/S. Médecine sciences [ISSN papier : 0767-0974 ; ISSN numérique : 1958-5381], 2002, Vol. 18, N° 2; p. 237-250 | fr_FR |
| dc.title | L’annotation in silico des séquences génomiques : Bio-informatique (1) | fr |
| dc.type | Article | fr_FR |
| dc.contributor.affiliation | URA 8030, Atelier de génomique comparative, Genoscope, 2, rue Gaston Crémieux, 91000 Évry, France | fr_FR |
| dc.contributor.affiliation | Institut InterUniversitaire Flamand de Biotechnologie (VIB), Département de Génétique Végétale, K.L. Ledeganckstraat, 35, 9000 Gand, Belgique | fr_FR |
| dc.identifier.doi | 10.1051/medsci/2002182237 | fr_FR |