



Trois gènes et quatre neuropathies périphériques myéliniques : premières corrélations génotype/phénotype
Résumé
Des anomalies des gènes codant pour PMP22, Cx32 et P0
sont responsables de la grande majorité des neuropathies
périphériques myéliniques, formant un continuum neuropathologique
: hypomyélinisation congénitale grave, syndrome
de Déjerine-Sottas, forme précoce et sévère, maladie de
Charcot-Marie-Tooth de type 1 ou liée à l’X, deux formes
moins sévères, neuropathie héréditaire avec paralysies à la
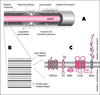
pression (HNPP) qui est la moins grave. La protéine P0 est
une protéine d’adhérence cellulaire, impliquée dans la compaction
de la myéline ; PMP22 est une protéine intégrale
membranaire, présente aussi dans la myéline compacte, à la
fonction encore inconnue ; la connexine Cx32 entre dans la
constitution des jonctions communicantes qui permettent la
diffusion radiale des molécules métaboliques et de signalisation
au travers des couches de la myéline. In most vertebrates, axons are usually ensheathed by myelin, a multi-lamellar structure that ensures the fidelity of nerve transmission and increases considerably nerve conduction velocity along the fibers. In the peripheral nervous system (PNS), myelin is formed by the extension of the plasma membrane of Schwann cells that wrap in spiral as many as 50 layers of double membrane structures around the axon. The myelin sheaths consist mostly of compact myelin that expresses a distinct set of structural proteins, namely myelin protein zero (P0), which is the most abundant component, peripheral myelin protein 22 (PMP22) and myelin basic protein. PNS compact myelin is interrupted by regions filled with cytoplasm, the incisures of Schmidt-Lanterman. These and the paranodal regions of Schwann cells express a distinct set of proteins that include myelin-associated glycoprotein and connexin32 (Cx32). It has now been demonstrated that genetic abnormalities in the genes encoding PMP22, P0 and Cx32, are responsible for the vast majority of demyelinating peripheral neuropathies, known as Charcot-Marie-Tooth disease type 1, X-linked Charcot-Marie-Tooth, Dejerine-Sottas syndrome, hereditary neuropathy with liability to pressure palsies and congenital hypomyelination. PMP22 is an integral membrane protein whose function is still poorly understood. P0 is a cell adhesion protein that contributes a sort of adhesive tape that holds together the extracellular leaflets of compact myelin. Cx32 is a channel-forming protein that is thought to provide the basis for a radial diffusional pathway of signaling molecules and metabolites across the myelin layers. Recent studies on the molecular structure and cell biology of these three pivotal proteins for myelin homeostasis have begun to shed light on some of the pathophysiological mechanisms that are specific to each syndrome. [References: 56]
Pour citer ce document
Pham-Dinh, D ; Blanquet-Grossard, F ; Ressot, C ; Bruzzone, R ; Dautigny, A, Trois gènes et quatre neuropathies périphériques myéliniques : premières corrélations génotype/phénotype, Med Sci (Paris), 1997, Vol. 13, N° 10; p.1113-22