| dc.contributor.author | de Verneuil, Hubert | - |
| dc.contributor.author | Robert-Richard, Elodie | - |
| dc.contributor.author | Ged, Cécile | - |
| dc.contributor.author | Mazurier, Frédéric | - |
| dc.contributor.author | Richard, Emmanuel | - |
| dc.contributor.author | Moreau-Gaudry, François | - |
| dc.date.accessioned | 2014-08-13T07:20:34Z | |
| dc.date.available | 2014-08-13T07:20:34Z | |
| dc.date.issued | 2008 | fr_FR |
| dc.identifier.citation | de Verneuil, Hubert ; Robert-Richard, Elodie ; Ged, Cécile ; Mazurier, Frédéric ; Richard, Emmanuel ; Moreau-Gaudry, François ; Succès de la thérapie génique d’un modèle murin de porphyrie érythropoïétique congénitale, Med Sci (Paris), 2008, Vol. 24, N° 6-7; p. 615-620 ; DOI : 10.1051/medsci/20082467615 | fr_FR |
| dc.identifier.issn | 1958-5381 | fr_FR |
| dc.identifier.uri | http://hdl.handle.net/10608/6477 | |
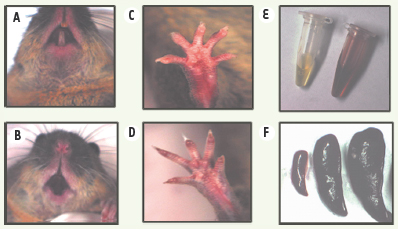
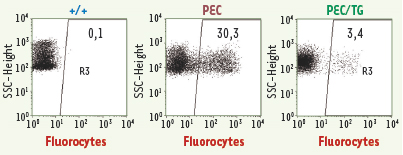
| dc.description.abstract | Les porphyries héréditaires représentent un ensemble de maladies métaboliques caractérisées par une synthèse, une accumulation et une excrétion accrues de porphyrines et/ou de leurs précurseurs, l’acide delta aminolévulinique et le porphobilinogène. Chacune de ces maladies a pu être reliée à un déficit spécifique d’une des enzymes de la biosynthèse de l’hème, et nous avons précédemment publié dans Médecine/Sciences les progrès effectués dans la connaissance des gènes, la pathologie moléculaire des porphyries ainsi que des modèles animaux indispensables pour des études physiopathologiques et thérapeutiques. Parmi les porphyries érythropoïétiques, la porphyrie érythropoïétique congénitale (PEC), ou maladie de Günther, la plus sévère des porphyries, est une maladie génétique caractérisée par un déficit en uroporphyrinogène III synthase (UROS). Elle est actuellement traitée par greffe de moelle osseuse allogénique dans les formes graves ; elle pourrait bénéficier dans le futur d’une thérapie génique ciblée sur les cellules souches/progénitrices hématopoïétiques. Les résultats d’une thérapie génique efficace dans un nouveau modèle murin de cette porphyrie sont exposés dans cet article. | fr |
| dc.description.abstract | Porphyrias are a group of disorders due to a genetic deficiency in one of the heme biosynthetic pathway enzymes. Congenital erythropoietic porphyria (CEP) is the most severe type characterized by a deficiency in uroporphyrinogen III synthase (UROS) activity. Bone marrow transplantation represents a curative treatment for patients, as long as human leucocyte antigen-compatible donor is available. We used a recently obtained murine model to check the feasibility of gene therapy in this disease. Lentivirus-mediated transfer of the human UROS cDNA into hematopoietic stem cells (HSCs) from Uros mut 248 mice resulted in a complete and long-term enzymatic, metabolic and phenotypic correction of the disease, favored by a survival advantage of corrected red blood cells. These results demonstrate for the first time that the cure of this mouse model of CEP at moderate transduction level supports the proof of concept of a gene therapy in this disease by transplantation of genetically modified HSCs. | en |
| dc.language.iso | fr | fr_FR |
| dc.publisher | EDK | fr_FR |
| dc.relation.ispartof | M/S revues | fr_FR |
| dc.rights | Article en libre accès | fr |
| dc.rights | Médecine/Sciences - Inserm - SRMS | fr |
| dc.source | M/S. Médecine sciences [ISSN papier : 0767-0974 ; ISSN numérique : 1958-5381], 2008, Vol. 24, N° 6-7; p. 615-620 | fr_FR |
| dc.subject.mesh | Animaux | fr |
| dc.subject.mesh | Modèles animaux de maladie humaine | fr |
| dc.subject.mesh | Thérapie génétique | fr |
| dc.subject.mesh | Humains | fr |
| dc.subject.mesh | Souris | fr |
| dc.subject.mesh | Porphyrie érythropoïétique congénitale | fr |
| dc.title | Succès de la thérapie génique d’un modèle murin de porphyrie érythropoïétique congénitale | fr |
| dc.title.alternative | Successful gene therapy of mice with congenital erythropoietic porphyria | en |
| dc.type | Article | fr_FR |
| dc.contributor.affiliation | Inserm, U876, Université Victor Segalen-Bordeaux 2, 146, rue Léo Saignat, 33076 Bordeaux Cedex, France | fr_FR |
| dc.identifier.doi | 10.1051/medsci/20082467615 | fr_FR |
| dc.identifier.pmid | 18601879 | fr_FR |